



Alexander Lucaci
@aglucaci.bsky.social
Postdoctoral Associate @ Weill Cornell Medicine.
PhD in Bioinformatics from Temple University
PhD in Bioinformatics from Temple University



Reposted by Alexander Lucaci
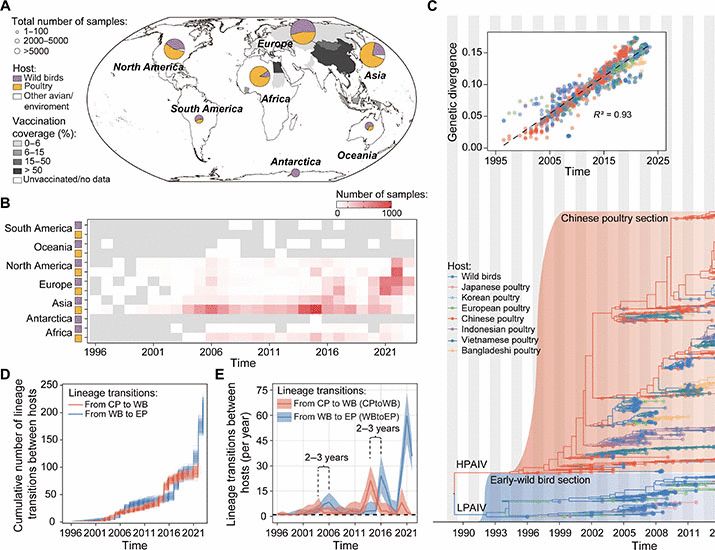
Building on the UShER tree of millions of SARS-CoV-2 genomes maintained by Angie Hinrichs, Hugh Haddox and Georg Angehrn (and others in @matsen.bsky.social lab and @jbloomlab.bsky.social) have looked into how the neutral mutation rate varies along the genome:
[1/N]
www.biorxiv.org/content/10.1...
[1/N]
www.biorxiv.org/content/10.1...

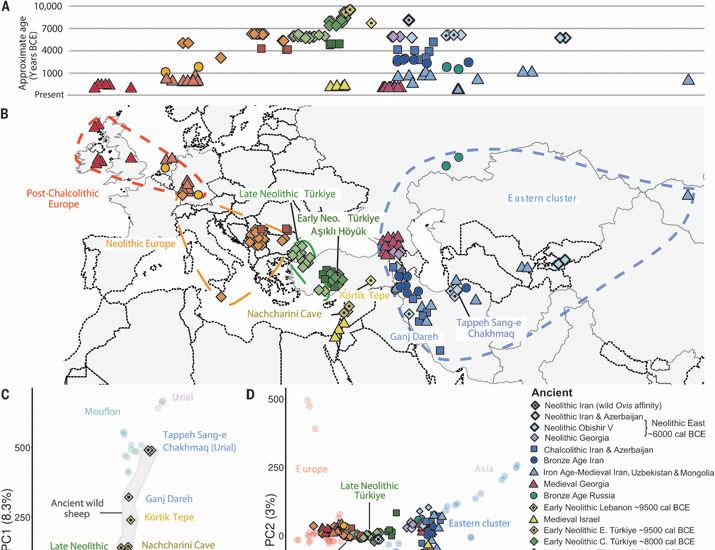
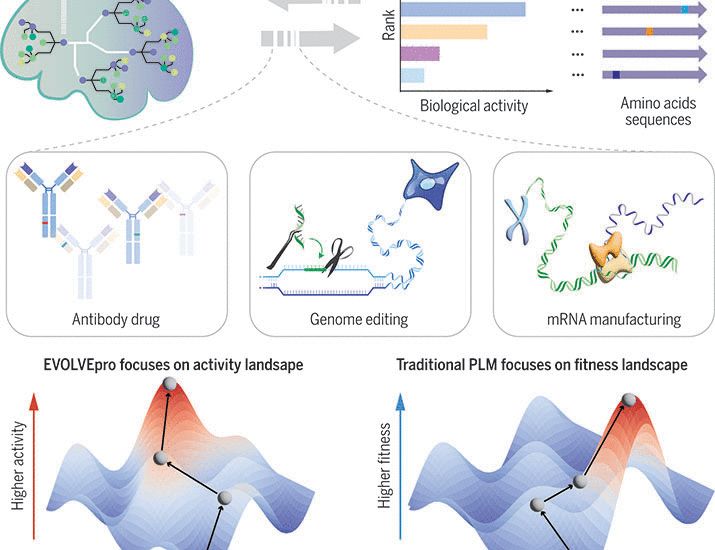
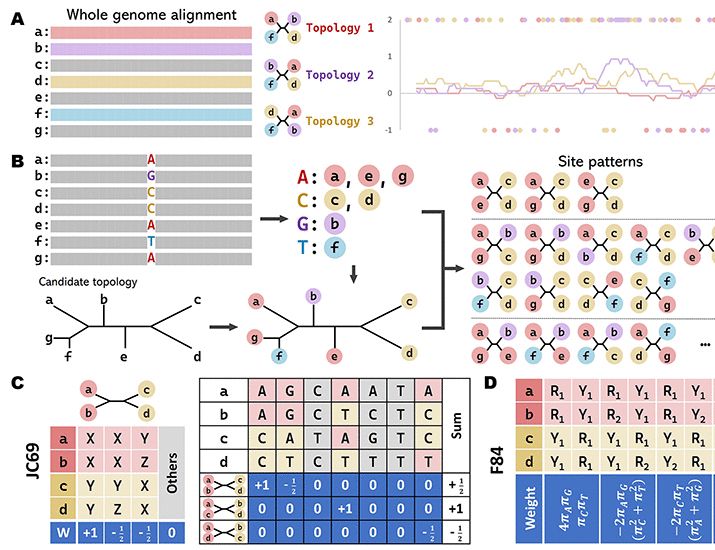
The mutation rate of SARS-CoV-2 is highly variable between sites and is influenced by sequence context, genomic region, and RNA structure
RNA viruses like SARS-CoV-2 have a high mutation rate, which contributes to their rapid evolution. The rate of mutations depends on the mutation type (e.g., A→C, A→G, etc.) and can vary between sites ...
www.biorxiv.org
January 11, 2025 at 5:37 PM
Building on the UShER tree of millions of SARS-CoV-2 genomes maintained by Angie Hinrichs, Hugh Haddox and Georg Angehrn (and others in @matsen.bsky.social lab and @jbloomlab.bsky.social) have looked into how the neutral mutation rate varies along the genome:
[1/N]
www.biorxiv.org/content/10.1...
[1/N]
www.biorxiv.org/content/10.1...
Reposted by Alexander Lucaci

Phylogenetic substitution rate estimates from millions-deep SARS-CoV-2 alignments

The mutation rate of SARS-CoV-2 is highly variable between sites and is influenced by sequence context, genomic region, and RNA structure https://www.biorxiv.org/content/10.1101/2025.01.07.631013v1
January 8, 2025 at 11:44 PM
Phylogenetic substitution rate estimates from millions-deep SARS-CoV-2 alignments
Reposted by Alexander Lucaci

Applications are now open for the 2025 Workshop on Molecular Evolution (MOLE) at the Marine Biological Laboratory in *beautiful* Woods Hole, MA!
More info here: molevolworkshop.github.io
Apply here: www.mbl.edu/education/ad...
Application deadline: Jan. 29th
Workshop dates: May 22nd - June 1st
More info here: molevolworkshop.github.io
Apply here: www.mbl.edu/education/ad...
Application deadline: Jan. 29th
Workshop dates: May 22nd - June 1st

Workshop on Molecular Evolution | Marine Biological Laboratory
The workshop serves graduate students, postdocs, and established faculty from around the world seeking to apply the principles of molecular evolution to questions of anthropology, conservation genetic...
www.mbl.edu
January 7, 2025 at 6:17 PM
Applications are now open for the 2025 Workshop on Molecular Evolution (MOLE) at the Marine Biological Laboratory in *beautiful* Woods Hole, MA!
More info here: molevolworkshop.github.io
Apply here: www.mbl.edu/education/ad...
Application deadline: Jan. 29th
Workshop dates: May 22nd - June 1st
More info here: molevolworkshop.github.io
Apply here: www.mbl.edu/education/ad...
Application deadline: Jan. 29th
Workshop dates: May 22nd - June 1st