
Ben Orr
@benorr.bsky.social
50 followers
110 following
9 posts
PhD Candidate, UCSF Biophysics, Kortemme Lab
Computational Biology & AI Lead, Animate Bio
ML for Protein Design
Posts
Media
Videos
Starter Packs
Reposted by Ben Orr


Ben Orr
@benorr.bsky.social
· Jun 10
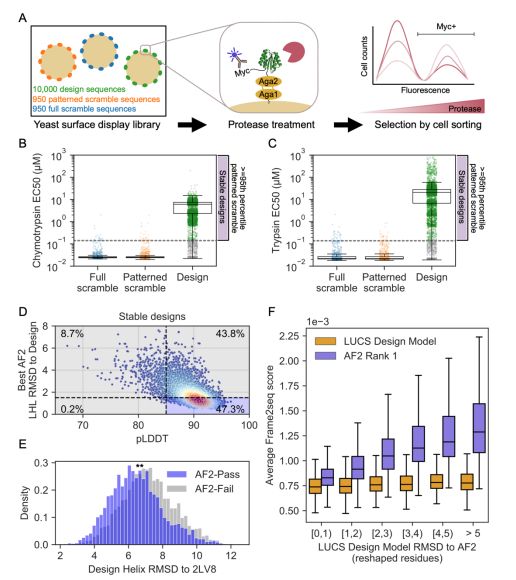
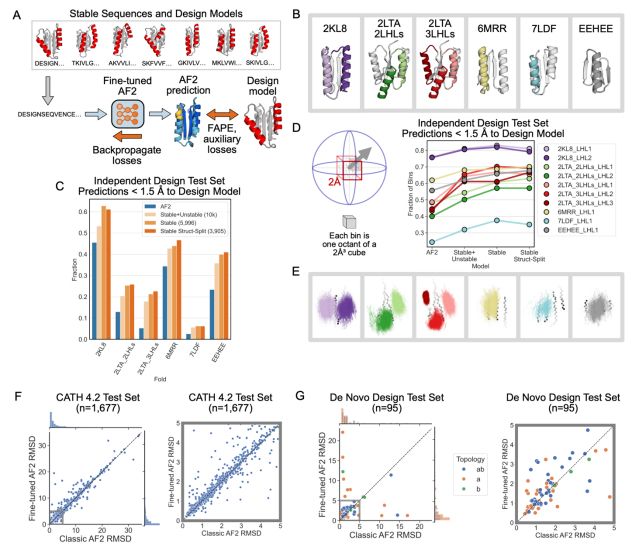
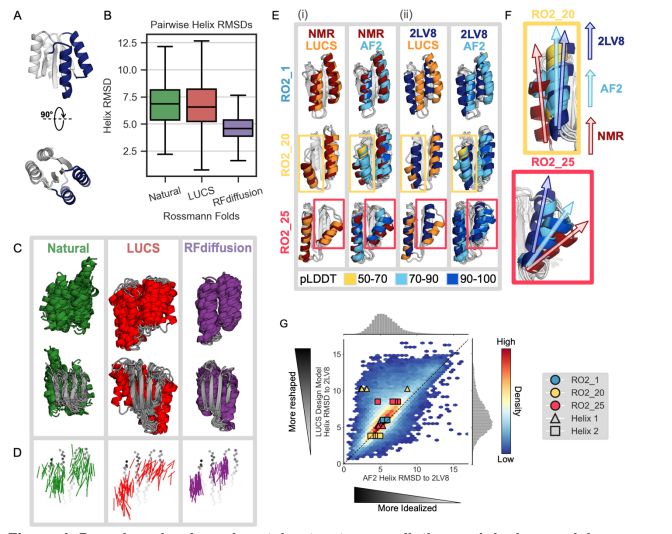
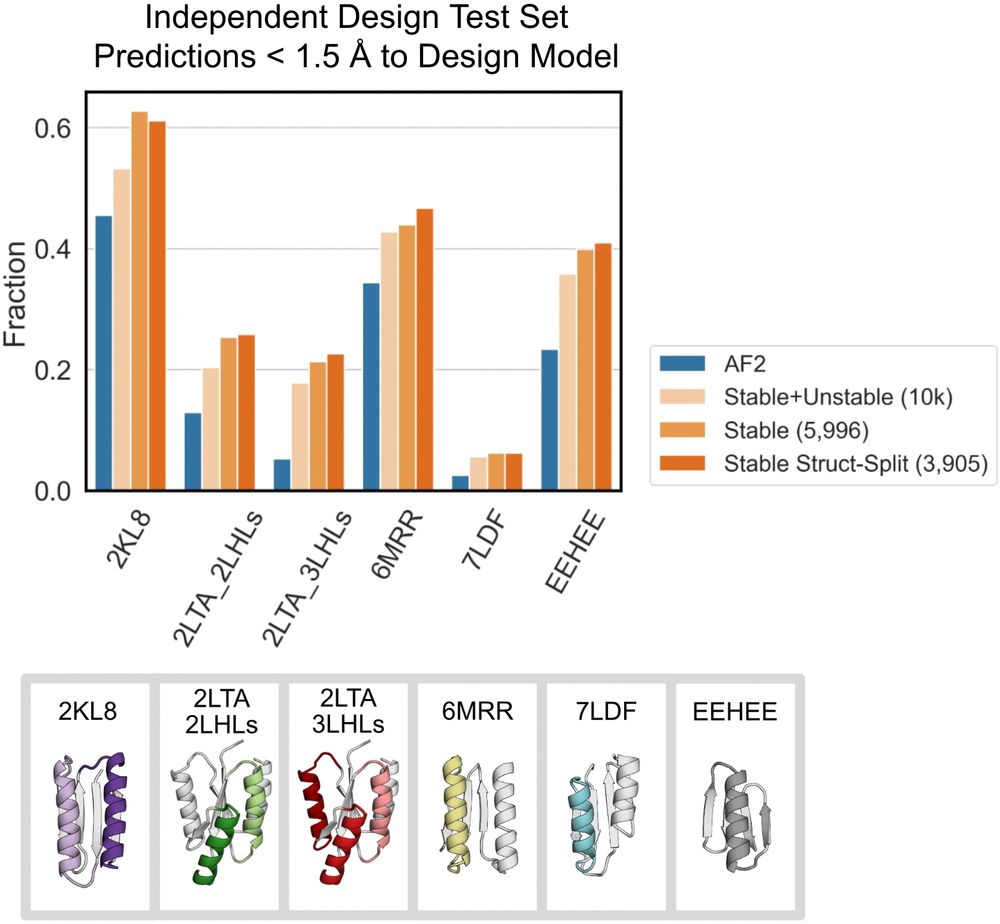
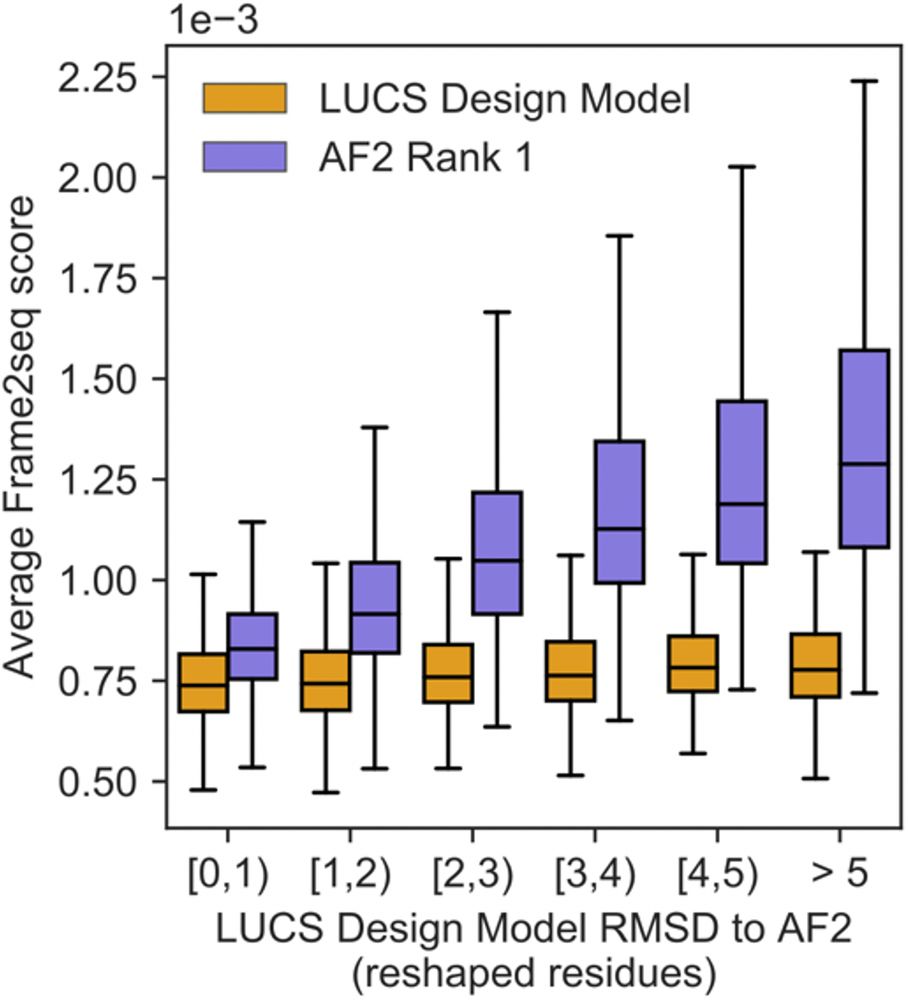
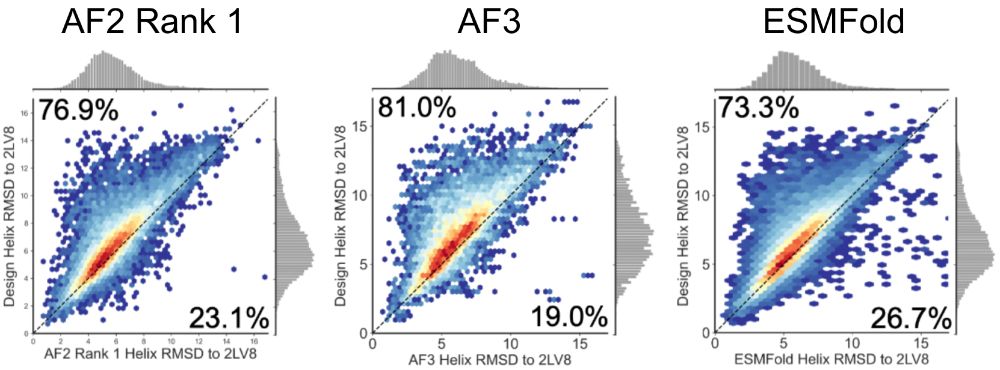
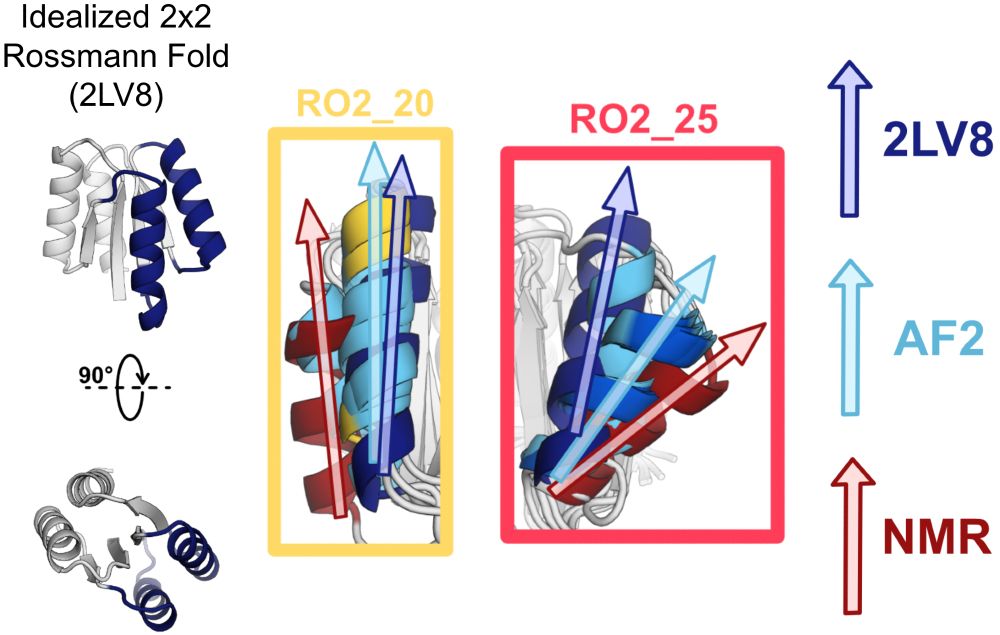
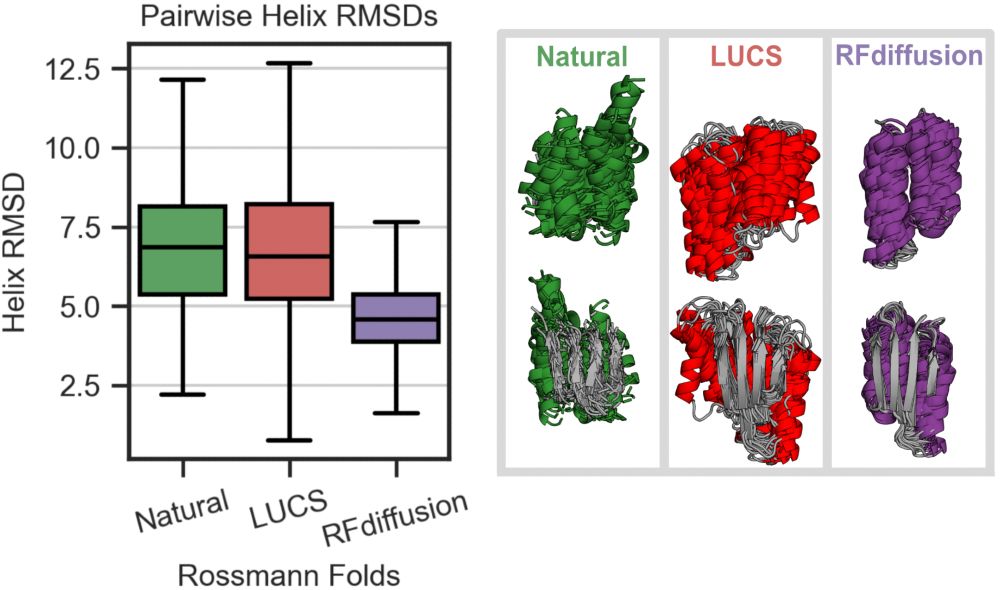
An improved model for prediction of de novo designed proteins with diverse geometries
Fine-tuned AF2 models for "Benjamin Orr*, Stephanie E. Crilly*, Deniz Akpinaroglu, Eleanor Zhu, Michael J. Keiser, Tanja Kortemme. An improved model for prediction of de novo designed proteins with di...
zenodo.org
Ben Orr
@benorr.bsky.social
· Jun 10
Ben Orr
@benorr.bsky.social
· Jun 10
Ben Orr
@benorr.bsky.social
· Jun 10

An improved model for prediction of de novo designed proteins with diverse geometries
Nature uses structural variations on protein folds to fine-tune the geometries of proteins for diverse functions, yet deep learning-based de novo protein design methods generate highly regular, ideali...
www.biorxiv.org