

Ceren Kilinc
@cerenkilinc.bsky.social
21 followers
46 following
1 posts
Phd candidate at Michigan State University, working with @dicksonlab.bsky.social
Interested in computational biochemistry, ML, MD, drug discovery
Obsessed with colors and art
Posts
Media
Videos
Starter Packs

Ceren Kilinc
@cerenkilinc.bsky.social
· Apr 8
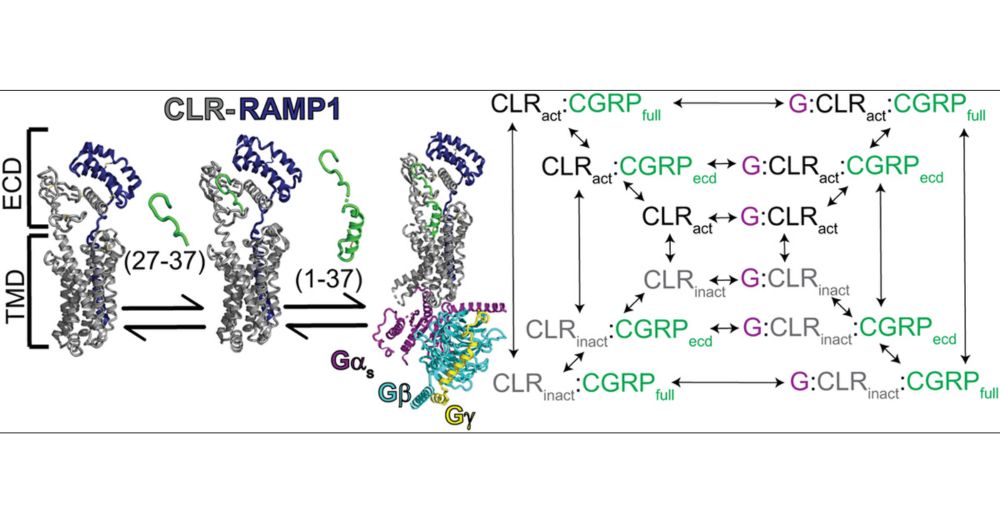
Characterization of the Two-Domain Peptide Binding Mechanism of the Human CGRP Receptor for CGRP and the Ultrahigh Affinity ssCGRP Variant
Calcitonin gene-related peptide (CGRP) is a 37-amino acid neuropeptide that functions in pain signaling and neuroimmune communication. The CGRP receptor, CGRPR, is a class B GPCR that is a drug target...
doi.org
Reposted by Ceren Kilinc

Samik Bose
@samikbose.bsky.social
· Feb 14

Markov State Models with Weighted Ensemble Simulation: How to Eliminate the Trajectory Merging Bias
The weighted ensemble (WE) algorithm is gaining popularity as a rare event method for studying long timescale processes with molecular dynamics. WE is particularly useful for determining kinetic properties, such as rates of protein (un)folding and ligand (un)binding, where transition rates can be calculated from the flux of trajectories into a target basin of interest. However, this flux depends exponentially on the number of splitting events that a given trajectory experiences before reaching the target state and can vary by orders of magnitude between WE replicates. Markov state models (MSMs) are helpful tools to aggregate information across multiple WE simulations and have previously been shown to provide more accurate transition rates than WE alone. Discrete-time MSMs are models that coarsely describe the evolution of the system from one discrete state to the next using a discrete lag time, τ. When an MSM is built using conventional MD data, longer values of τ typically provide more accurate results. Combining WE simulations with Markov state modeling presents some additional challenges, especially when using a value of τ that exceeds the lag time between resampling steps in the WE algorithm, τWE. Here, we identify a source of bias that occurs when τ > τWE, which we refer to as “merging bias”. We also propose an algorithm to eliminate the merging bias, which results in merging bias-corrected MSMs, or “MBC-MSMs”. Using a simple model system, as well as a complex biomolecular example, we show that MBC-MSMs significantly outperform both τ = τWE MSMs and uncorrected MSMs at longer lag times.
doi.org