

Gregg Thomas
@gwct.bio
370 followers
320 following
9 posts
Bioinformatics Scientist at Harvard FAS Informatics. Evolution, Genomics, Phylogenetics. Dog walker. He/him. gwct.bio // informatics.fas.harvard.edu
Posts
Media
Videos
Starter Packs
Reposted by Gregg Thomas

Fábio K. Mendes
@fabiology.bsky.social
· Jul 12

Gregg Thomas
@gwct.bio
· Jun 4

GitHub - harvardinformatics/snakemake-executor-plugin-cannon: A Snakemake executor plugin for submitting jobs to the Harvard Cannon cluster
A Snakemake executor plugin for submitting jobs to the Harvard Cannon cluster - harvardinformatics/snakemake-executor-plugin-cannon
github.com
Gregg Thomas
@gwct.bio
· Jun 4

GitHub - harvardinformatics/cactus-snakemake: Snakemake workflows for performing whole genome alignment with Cactus efficiently on SLURM clusters
Snakemake workflows for performing whole genome alignment with Cactus efficiently on SLURM clusters - harvardinformatics/cactus-snakemake
github.com
Reposted by Gregg Thomas


Reposted by Gregg Thomas


Reposted by Gregg Thomas


Reposted by Gregg Thomas



Gregg Thomas
@gwct.bio
· Dec 4
Gregg Thomas
@gwct.bio
· Dec 4
Gregg Thomas
@gwct.bio
· Dec 4


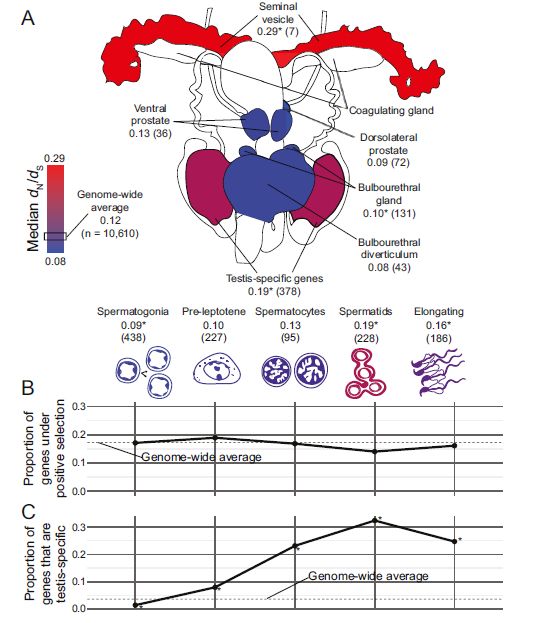

Reposted by Gregg Thomas

Jeremy BOO! Yoder 🎃💀🕷️
@jbyoder.org
· Oct 28
Reposted by Gregg Thomas

Reposted by Gregg Thomas
Matthew Hahn
@3rdreviewer.bsky.social
· Oct 18

Multiple, Tenured/Tenure-Track Faculty Positions in Computing in Biology, Health, and Wellness - Bloomington, Indiana job with Indiana University Bloomington | 664327
Invite applications for multiple tenure /tenure-track positions at the Assistant, Associate, or Full Professor ranks
jobs.sciencecareers.org
Reposted by Gregg Thomas

Reposted by Gregg Thomas



Reposted by Gregg Thomas
Nat Forsdick
@natforsdick.bsky.social
· Jul 27
Reposted by Gregg Thomas
Matthew Hahn
@3rdreviewer.bsky.social
· Jan 3