

Anthony Nash PhD
@anthonyc1nash.bsky.social
50 followers
52 following
53 posts
Computational Chemist. Theoretical Biophysicist (physics-based modelling). Protein Dynamics. Unconventional Computing. Metalloproteases.
Posts
Media
Videos
Starter Packs

Reposted by Anthony Nash PhD



Reposted by Anthony Nash PhD



Reposted by Anthony Nash PhD

Johannes Gorges
@jogorges.bsky.social
· Sep 8
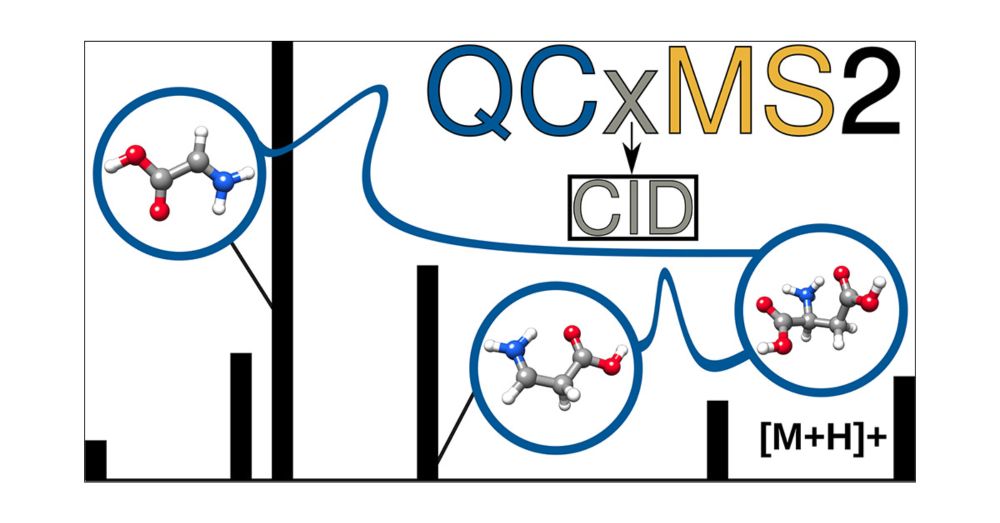
Evaluation of the QCxMS2 Method for the Calculation of Collision-Induced Dissociation Spectra via Automated Reaction Network Exploration
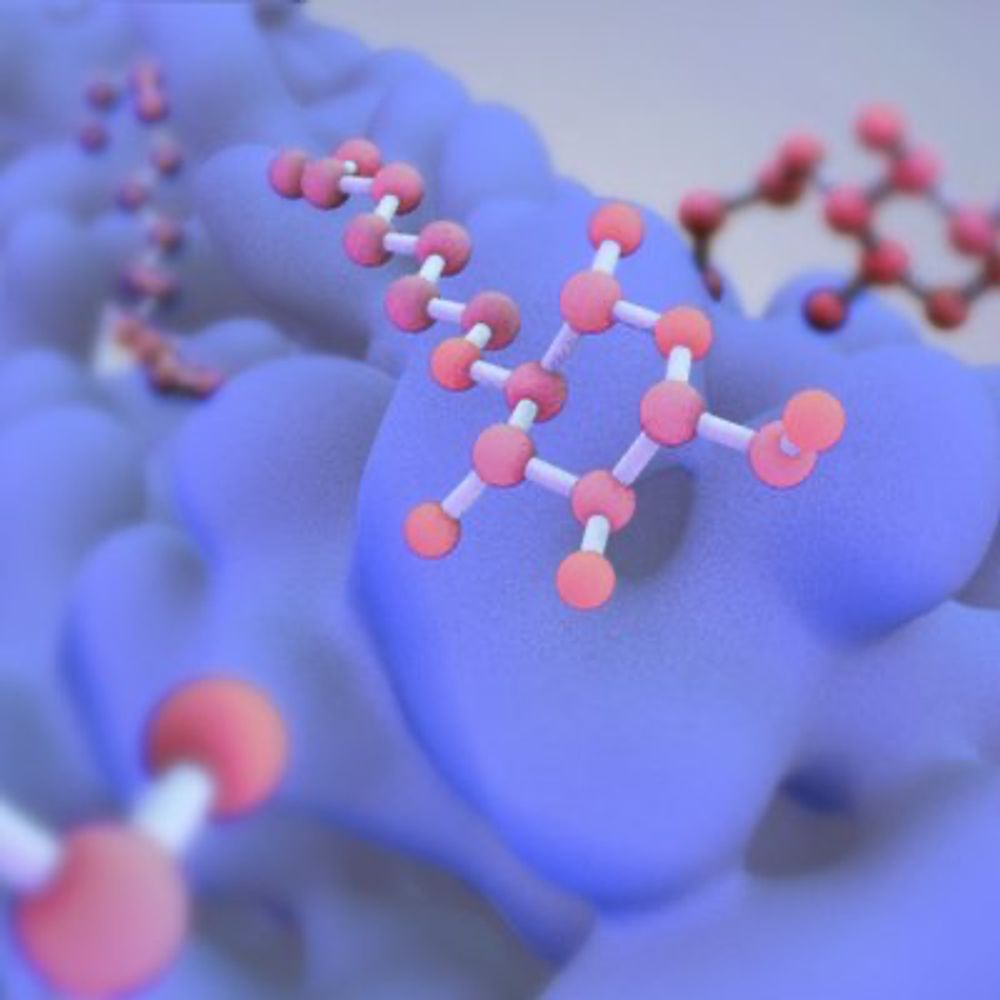
Collision-induced dissociation mass spectrometry (CID-MS) is an important tool in analytical chemistry for the structural elucidation of unknown compounds. The theoretical prediction of the CID spectra plays a critical role in supporting and accelerating this process. To this end, we adapt the recently developed QCxMS2 program originally designed for the calculation of electron ionization (EI) spectra to enable the computation of CID-MS. To account for the fragmentation conditions characteristic of CID within the automated reaction network discovery approach of QCxMS2 we adapted the internal energy distribution to match the experimental conditions. This distribution can be adjusted via a single parameter to approximate various activation settings, thereby eliminating the need for explicit simulations of the collisional process. We evaluate our approach on a test set of 13 organic molecules with diverse functional groups, compiled specifically for this study. All reference spectra were recorded consistently under the same measurement conditions, including both CID and higher-energy collisional dissociation (HCD) modes. Overall, QCxMS2 achieves a good average entropy similarity score (ESS) of 0.687 for the HCD spectra and 0.773 for the CID spectra. The direct comparison to experimental data demonstrates that the QCxMS2 approach, even without explicit modeling of collisions, is generally capable of computing both CID and HCD spectra with reasonable accuracy and robustness. This highlights its potential as a valuable tool for integration into structure elucidation workflows in analytical mass spectrometry.
doi.org






Reposted by Anthony Nash PhD



Reposted by Anthony Nash PhD

Cole Group
@colegroupncl.bsky.social
· May 19

MACE-OFF: Short-Range Transferable Machine Learning Force Fields for Organic Molecules
Classical empirical force fields have dominated biomolecular simulations for over 50 years. Although widely used in drug discovery, crystal structure prediction, and biomolecular dynamics, they generally lack the accuracy and transferability required for first-principles predictive modeling. In this paper, we introduce MACE-OFF, a series of short-range transferable force fields for organic molecules created using state-of-the-art machine learning technology and first-principles reference data computed with a high level of quantum mechanical theory. MACE-OFF demonstrates the remarkable capabilities of short-range models by accurately predicting a wide variety of gas- and condensed-phase properties of molecular systems. It produces accurate, easy-to-converge dihedral torsion scans of unseen molecules as well as reliable descriptions of molecular crystals and liquids, including quantum nuclear effects. We further demonstrate the capabilities of MACE-OFF by determining free energy surfaces in explicit solvent as well as the folding dynamics of peptides and nanosecond simulations of a fully solvated protein. These developments enable first-principles simulations of molecular systems for the broader chemistry community at high accuracy and relatively low computational cost.
pubs.acs.org

Reposted by Anthony Nash PhD

