



@allydunham.bsky.social
Reposted

New 🧬✂️ pre-print! We show that paired prime editing can efficiently generate large deletions — even >1 Mb — with high precision and at scale. We use this to perform the first pooled prime deletion screen across the human genome.
🔗 biorxiv.org/content/10.1...
A short thread (by Juliane Weller)👇
🔗 biorxiv.org/content/10.1...
A short thread (by Juliane Weller)👇

Generating long deletions across the genome with pooled paired prime editing screens
Engineered deletions are a powerful probe for studying genome architecture, function, and regulation. Yet, the lack of effective methods to create them in large numbers and at multi-kilobase scale has...
biorxiv.org
November 5, 2025 at 2:17 PM
New 🧬✂️ pre-print! We show that paired prime editing can efficiently generate large deletions — even >1 Mb — with high precision and at scale. We use this to perform the first pooled prime deletion screen across the human genome.
🔗 biorxiv.org/content/10.1...
A short thread (by Juliane Weller)👇
🔗 biorxiv.org/content/10.1...
A short thread (by Juliane Weller)👇
We've been screening across many DNA construct structures with variable number/size of interesting regions - e.g. spacer, extension and barcode for prime editing. There wasn't a widely used tool to process these complex structured reads so I ended up developing github.com/allydunham/d....

GitHub - allydunham/dnacomb: CLI tool for flexibly parsing structured sequence reads into count tables and comparing them to expected libraries
CLI tool for flexibly parsing structured sequence reads into count tables and comparing them to expected libraries - allydunham/dnacomb
github.com
October 13, 2025 at 2:36 PM
We've been screening across many DNA construct structures with variable number/size of interesting regions - e.g. spacer, extension and barcode for prime editing. There wasn't a widely used tool to process these complex structured reads so I ended up developing github.com/allydunham/d....
Reposted

MPRAbase (mprabase.ucsf.edu) , a customized database for massively parallel reporter assays (MPRAs) to easily find and download MPRA data. Amazing work by Jingjing Zhao, Fotis Baltoumas, Georgios Pavlopoulos, @vagar.bsky.social, ilias Georgakopoulos-Soares & others.
genome.cshlp.org/content/earl...
genome.cshlp.org/content/earl...
MPRAbase a Massively Parallel Reporter Assay database
An international, peer-reviewed genome sciences journal featuring outstanding original research that offers novel insights into the biology of all organisms
genome.cshlp.org
April 22, 2025 at 8:51 PM
MPRAbase (mprabase.ucsf.edu) , a customized database for massively parallel reporter assays (MPRAs) to easily find and download MPRA data. Amazing work by Jingjing Zhao, Fotis Baltoumas, Georgios Pavlopoulos, @vagar.bsky.social, ilias Georgakopoulos-Soares & others.
genome.cshlp.org/content/earl...
genome.cshlp.org/content/earl...
Reposted
We're hiring to expand on the work to understand the human genome by engineering it!
lnkd.in/da-gitNc
lnkd.in/da-gitNc
February 10, 2025 at 8:53 AM
We're hiring to expand on the work to understand the human genome by engineering it!
lnkd.in/da-gitNc
lnkd.in/da-gitNc
Reposted
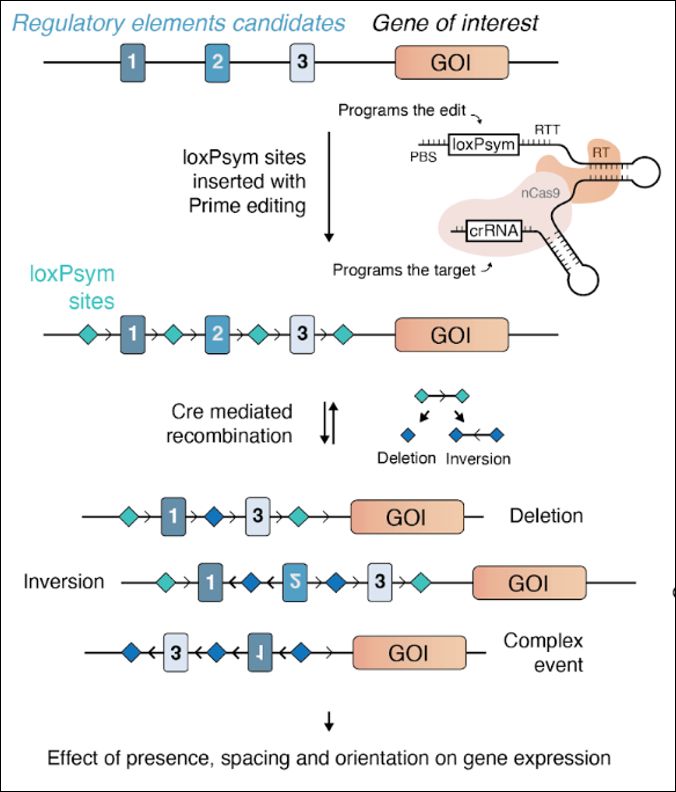
We are happy to share our enhancer scramble story, a strategy to create hundreds of stochastic deletions, inversions, and duplications within mammalian gene regulatory regions and associate these new architectures with gene expression levels 🧵
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
January 15, 2025 at 8:32 PM
We are happy to share our enhancer scramble story, a strategy to create hundreds of stochastic deletions, inversions, and duplications within mammalian gene regulatory regions and associate these new architectures with gene expression levels 🧵
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted
Does my mutation have the same impact as yours? Population genetics 🤠 🥸 🤓 🤡 meets single cell CRISPRi ⚡ ! www.biorxiv.org/content/10.1... Led by Claudia Feng, Oliver Stegle, Britta Velten, @sangerinstitute.bsky.social .
LinkedIn
This link will take you to a page that’s not on LinkedIn
lnkd.in
December 2, 2024 at 1:57 PM
Does my mutation have the same impact as yours? Population genetics 🤠 🥸 🤓 🤡 meets single cell CRISPRi ⚡ ! www.biorxiv.org/content/10.1... Led by Claudia Feng, Oliver Stegle, Britta Velten, @sangerinstitute.bsky.social .