



Chao Hou
@chaohou.bsky.social
Protein dynamic, Multi conformation, Language model, Computational biology | Postdoc @Columbia | PhD 2023 & Bachelor 2020 @PKU1898
http://chaohou.netlify.app
http://chaohou.netlify.app
We just updated our manuscript "Understanding Language Model Scaling on Protein Fitness Prediction". Where we explained why larger pLMs don’t always perform better on mutation effect prediction. We extended beyond ESM2 to models like ESMC, ESM3, SaProt, and ESM-IF1.
#ProteinLM
#ProteinLM

August 25, 2025 at 2:25 AM
We just updated our manuscript "Understanding Language Model Scaling on Protein Fitness Prediction". Where we explained why larger pLMs don’t always perform better on mutation effect prediction. We extended beyond ESM2 to models like ESMC, ESM3, SaProt, and ESM-IF1.
#ProteinLM
#ProteinLM
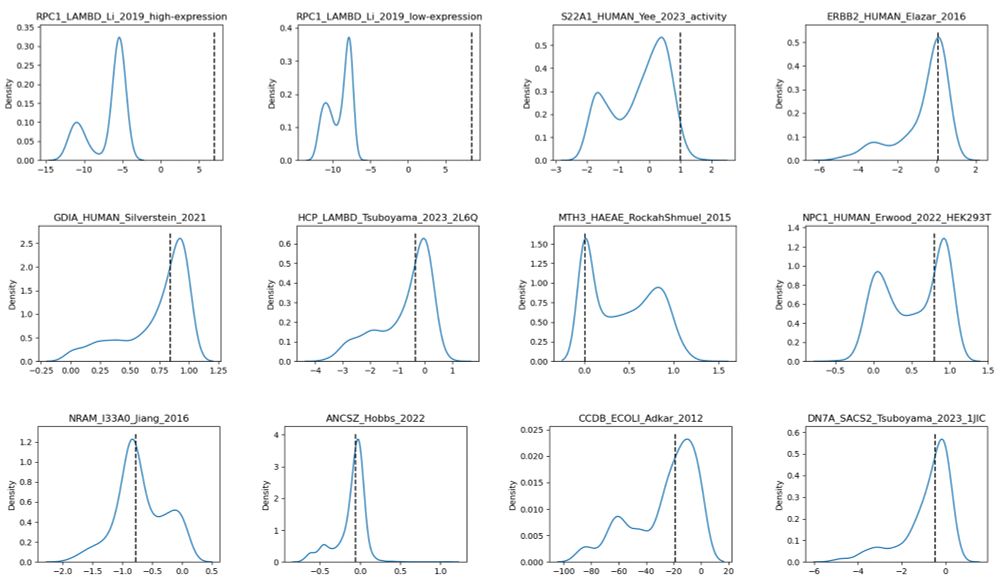
⚠️ Caution when using ProteinGYM binary classification: some DMS_binarization_cutoff values appear inverted, some are totally unreasonable given the DMS_score shows a clear bimodal distribution.
June 11, 2025 at 3:57 PM
⚠️ Caution when using ProteinGYM binary classification: some DMS_binarization_cutoff values appear inverted, some are totally unreasonable given the DMS_score shows a clear bimodal distribution.
Reposted by Chao Hou

How can we better understand pathogenic variants in intrinsically disordered regions (IDRs)? How do models such as AlphaMissense and ESM1b predict pathogenicity, when these regions typically exhibit lower genomic conservation than ordered regions? Read more:
doi.org/10.1101/2025...
doi.org/10.1101/2025...

Molecular dynamics simulations of intrinsically disordered protein regions enable biophysical interpretation of variant effect predictors
Predictive models for missense variant pathogenicity offer little functional interpretation for intrinsically disordered regions, since they rely on conservation and coevolution across homologous sequ...
doi.org
May 13, 2025 at 2:15 PM
How can we better understand pathogenic variants in intrinsically disordered regions (IDRs)? How do models such as AlphaMissense and ESM1b predict pathogenicity, when these regions typically exhibit lower genomic conservation than ordered regions? Read more:
doi.org/10.1101/2025...
doi.org/10.1101/2025...
Reposted by Chao Hou

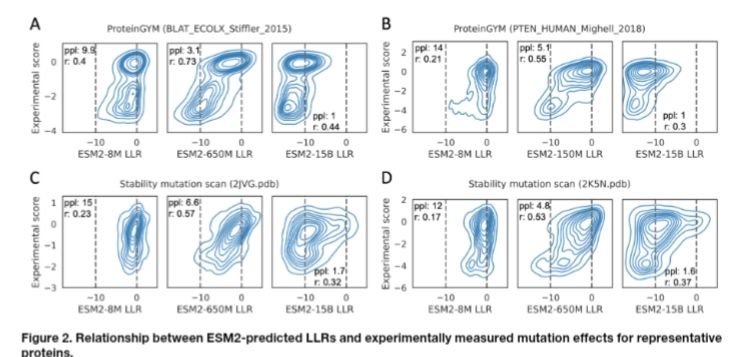
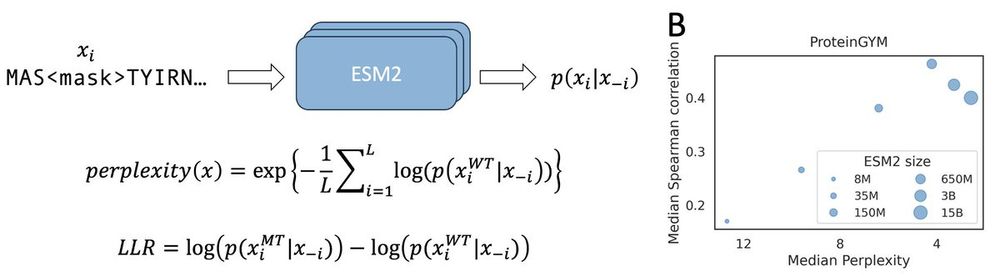
Protein language model likelihood are better zero shot mutation effect predictions when they have perplexity 3-6 on the wildtype sequence.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
April 30, 2025 at 6:18 PM
Protein language model likelihood are better zero shot mutation effect predictions when they have perplexity 3-6 on the wildtype sequence.
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
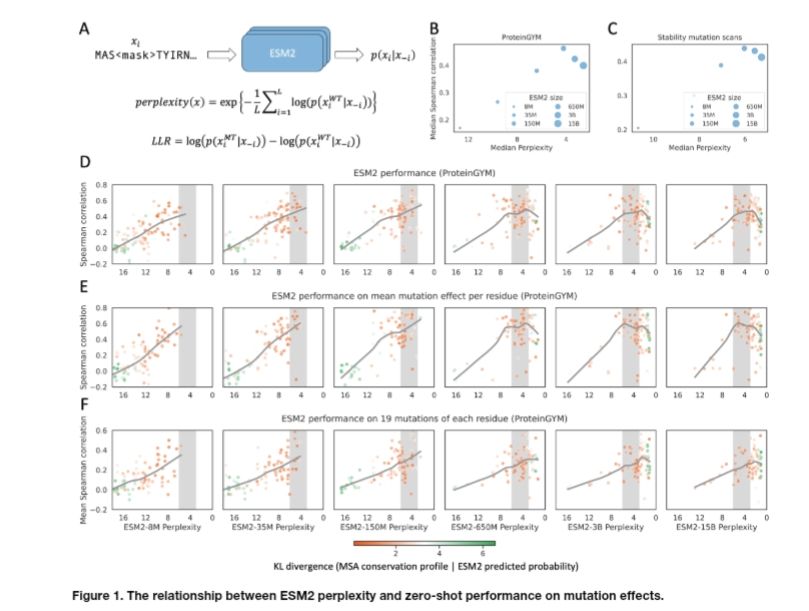
Why do large protein language models like ESM2-15B underperform compared to medium-sized ones like ESM2-650M in predicting mutation effects? 🤔
We dive into this issue in our new preprint—bringing insights into model scaling on mutation effect prediction. 🧬📉
We dive into this issue in our new preprint—bringing insights into model scaling on mutation effect prediction. 🧬📉
April 29, 2025 at 5:54 PM
Why do large protein language models like ESM2-15B underperform compared to medium-sized ones like ESM2-650M in predicting mutation effects? 🤔
We dive into this issue in our new preprint—bringing insights into model scaling on mutation effect prediction. 🧬📉
We dive into this issue in our new preprint—bringing insights into model scaling on mutation effect prediction. 🧬📉
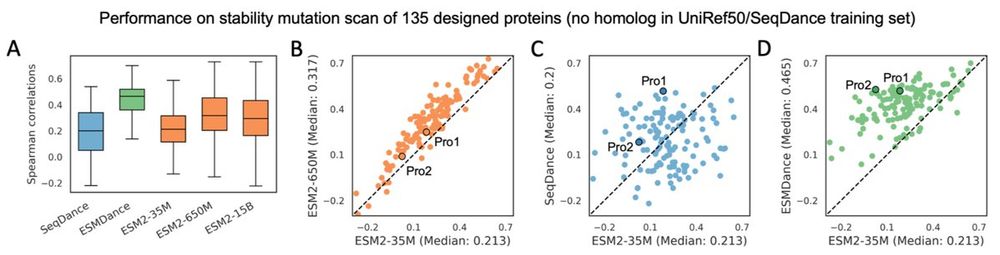
We have updated our protein lanuage model trained on structure dynamics. Our new models show significant better zero-shot performance on mutation effects of designed and viral proteins compared to ESM2. check the new preprint here: www.biorxiv.org/content/10.1...
April 17, 2025 at 2:40 PM
We have updated our protein lanuage model trained on structure dynamics. Our new models show significant better zero-shot performance on mutation effects of designed and viral proteins compared to ESM2. check the new preprint here: www.biorxiv.org/content/10.1...
Reposted by Chao Hou

SeqDance: A Protein Language Model for Representing Protein Dynamic Properties https://www.biorxiv.org/content/10.1101/2024.10.11.617911v1

SeqDance: A Protein Language Model for Representing Protein Dynamic Properties https://www.biorxiv.org/content/10.1101/2024.10.11.617911v1
Proteins perform their functions by folding amino acid sequences into dynamic structural ensembles.
www.biorxiv.org
October 15, 2024 at 4:49 PM
SeqDance: A Protein Language Model for Representing Protein Dynamic Properties https://www.biorxiv.org/content/10.1101/2024.10.11.617911v1