



Chris Saunders
@ctsa.bsky.social
Rare disease and cancer analysis models for sequencing data. Scientist at PacBio. Art school survivor. Views my own.


Reposted by Chris Saunders

I am hiring a staff bioinformatician for my new lab at the University of Utah! Please consider applying if you are on the hunt:
employment.utah.edu/salt-lake-ci...
employment.utah.edu/salt-lake-ci...
Jobs | University of Utah
Founded in 1850, The University of Utah is the flagship institution of higher learning in Utah, and offers over 100 undergraduate and more than 90 graduate degree programs to over 30,000 students. Uni...
employment.utah.edu
January 12, 2026 at 9:04 PM
I am hiring a staff bioinformatician for my new lab at the University of Utah! Please consider applying if you are on the hunt:
employment.utah.edu/salt-lake-ci...
employment.utah.edu/salt-lake-ci...
Reposted by Chris Saunders

Absolutely thrilled to share the latest work from my lab focused on the variation and evolution of human centromeres among global populations! We assembled 2,110 human centromeres, identifying 226 new major haplotypes and 1,870 α-satellite HOR variants. www.biorxiv.org/content/10.6...

December 16, 2025 at 4:06 PM
Absolutely thrilled to share the latest work from my lab focused on the variation and evolution of human centromeres among global populations! We assembled 2,110 human centromeres, identifying 226 new major haplotypes and 1,870 α-satellite HOR variants. www.biorxiv.org/content/10.6...
Reposted by Chris Saunders

Join us at this exciting Workshop on #LongReads with keynotes from @aphillippy.bsky.social @mydennis.bsky.social
co-organized by @christinebeck.bsky.social and Mark Adams @jacksonlab.bsky.social Register www.jax.org/longread
#RNA #genomics #bioinformatics #cancer #medicine #evolution #neuroscience
co-organized by @christinebeck.bsky.social and Mark Adams @jacksonlab.bsky.social Register www.jax.org/longread
#RNA #genomics #bioinformatics #cancer #medicine #evolution #neuroscience

Long-Read Sequencing Workshop
The program will involve sessions on Genome Evolution, Genome Regulation, Transcriptomics, Rare Diseases, Common Diseases, and Viral and Microbial Genomes. Committed speakers include Adam Phillipy (NH...
www.jax.org
November 14, 2025 at 7:57 PM
Join us at this exciting Workshop on #LongReads with keynotes from @aphillippy.bsky.social @mydennis.bsky.social
co-organized by @christinebeck.bsky.social and Mark Adams @jacksonlab.bsky.social Register www.jax.org/longread
#RNA #genomics #bioinformatics #cancer #medicine #evolution #neuroscience
co-organized by @christinebeck.bsky.social and Mark Adams @jacksonlab.bsky.social Register www.jax.org/longread
#RNA #genomics #bioinformatics #cancer #medicine #evolution #neuroscience
Reposted by Chris Saunders

My new tool Paraviewer is now available for use at github.com/PacificBiosc...! If you use Paraphase, try this new next-step tool - it automates and greatly simplifies variant visualization from Paraphase variant calling. If you're at #ASHG2025, visit me today at poster 4109W. #pacbio

GitHub - PacificBiosciences/Paraviewer
Contribute to PacificBiosciences/Paraviewer development by creating an account on GitHub.
github.com
October 15, 2025 at 2:58 PM
My new tool Paraviewer is now available for use at github.com/PacificBiosc...! If you use Paraphase, try this new next-step tool - it automates and greatly simplifies variant visualization from Paraphase variant calling. If you're at #ASHG2025, visit me today at poster 4109W. #pacbio
Reposted by Chris Saunders
My complex variant visualization tool SVTopo is now officially published in BMC Genomics! link.springer.com/article/10.1.... This tool allows HiFi users to view complex germline structural variation in intuitive and informative plots.

Complex structural variant visualization with SVTopo - BMC Genomics
Background Structural variants are genomic variants that impact at least 50 nucleotides. Structural variants can play major roles in diversity and human health. Many structural variants are difficult to interpret and understand with existing visualization tools, especially when comprised of inverted sequences or multiple breakend pairs. Results We present SVTopo, a tool to visualize germline structural variants with supporting evidence from high-accuracy long reads in easily understood figures. We include examples of 101 visually complex structural variants from seven unrelated human genomes, manually assigned to ten categories. These demonstrate a broad spectrum of rearrangement and showcase the frequency of complex structural variants in human genomes. Conclusions SVTopo shows breakpoint evidence in ways that aid reasoning about the impact of multi-breakpoint rearrangements. The images created aid human reasoning about the result of structural variation on gene and regulatory regions.
link.springer.com
October 9, 2025 at 7:00 PM
My complex variant visualization tool SVTopo is now officially published in BMC Genomics! link.springer.com/article/10.1.... This tool allows HiFi users to view complex germline structural variation in intuitive and informative plots.
Reposted by Chris Saunders

New pre-print from the Banfield lab, highlighting an interesting case of 1.5Mb megaplasmids found in human gut.
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...

Megaplasmids associate with Escherichia coli and other Enterobacteriaceae
Humans and animals are ubiquitously colonized by Enterobacteriaceae , a bacterial family that contains both commensals and clinically significant pathogens. Here, we report Enterobacteriaceae megaplas...
doi.org
October 1, 2025 at 4:44 PM
New pre-print from the Banfield lab, highlighting an interesting case of 1.5Mb megaplasmids found in human gut.
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...
Reposted by Chris Saunders

I'm excited to share our pre-print about a new variant benchmarking tool we've been working on for the past few months!
Aardvark: Sifting through differences in a mound of variants
GitHub: github.com/PacificBiosc...
Some highlights in this thread:
1/N
Aardvark: Sifting through differences in a mound of variants
GitHub: github.com/PacificBiosc...
Some highlights in this thread:
1/N
October 6, 2025 at 8:07 PM
I'm excited to share our pre-print about a new variant benchmarking tool we've been working on for the past few months!
Aardvark: Sifting through differences in a mound of variants
GitHub: github.com/PacificBiosc...
Some highlights in this thread:
1/N
Aardvark: Sifting through differences in a mound of variants
GitHub: github.com/PacificBiosc...
Some highlights in this thread:
1/N
In Stockholm for work and got a great recommendation to Klättercentret Telefonplan for a boulder break last night. Great gym all around and easy hop on the metro from city center.


October 5, 2025 at 6:25 AM
In Stockholm for work and got a great recommendation to Klättercentret Telefonplan for a boulder break last night. Great gym all around and easy hop on the metro from city center.
Reposted by Chris Saunders

Congrats to @dantipov.bsky.social et al. on the publication of Verkko2! The team put a ton of work into this making it the first assembler that deals with the complexity of human acrocentric chromosomes. Lots of interesting discoveries to come! genome.cshlp.org/content/earl...

June 17, 2025 at 1:39 PM
Congrats to @dantipov.bsky.social et al. on the publication of Verkko2! The team put a ton of work into this making it the first assembler that deals with the complexity of human acrocentric chromosomes. Lots of interesting discoveries to come! genome.cshlp.org/content/earl...
Reposted by Chris Saunders

Now published! Note that since Vikram's original post (quoted here), he's made it easy to dynamically update a set of multi-MUMs (e.g. when more genomes are added to a pangenome) and to find multi-MUMs for huge collections like HPRCv2 genomebiology.biomedcentral.com/articles/10....
June 17, 2025 at 2:02 PM
Now published! Note that since Vikram's original post (quoted here), he's made it easy to dynamically update a set of multi-MUMs (e.g. when more genomes are added to a pangenome) and to find multi-MUMs for huge collections like HPRCv2 genomebiology.biomedcentral.com/articles/10....
Reposted by Chris Saunders
Very excited to share I’ll be starting my own group at University of Utah in the Department of Human Genetics in the new year! Reach out if you are interested! vollgerlab.com
June 5, 2025 at 2:15 PM
Very excited to share I’ll be starting my own group at University of Utah in the Department of Human Genetics in the new year! Reach out if you are interested! vollgerlab.com
Reposted by Chris Saunders

Just released a major update to the sawfish SV caller, which adds CNV calling and integration. Assessment on a set of pathogenic CNVs from Gross et al. shows this integrated-call strategy can substantially improve (single-method) recall, especially at lower HiFi sequencing depths

May 20, 2025 at 12:52 AM
Just released a major update to the sawfish SV caller, which adds CNV calling and integration. Assessment on a set of pathogenic CNVs from Gross et al. shows this integrated-call strategy can substantially improve (single-method) recall, especially at lower HiFi sequencing depths
Reposted by Chris Saunders

Long-read sequencing of large pedigrees is an ideal way to map all classes of denovo mutations! A collaboration between University of Utah, University of Washington, and PacBio. Glad to be a part of this project 👏
www.nature.com/articles/s41...
www.nature.com/articles/s41...

Human de novo mutation rates from a four-generation pedigree reference - Nature
Analysis of more than 95% of each diploid human genome of a four-generation, twenty-eight-member family using five complementary short-read and long-read sequencing technologies provides a truth set t...
www.nature.com
April 23, 2025 at 4:11 PM
Long-read sequencing of large pedigrees is an ideal way to map all classes of denovo mutations! A collaboration between University of Utah, University of Washington, and PacBio. Glad to be a part of this project 👏
www.nature.com/articles/s41...
www.nature.com/articles/s41...
Reposted by Chris Saunders
This example is hard to understand from e.g. IGV/Ribbon (see Fig1) but pretty simple in SVTopo: 4 blocks deleted (B,D,F,H), 2 inverted (E,G), 1 re-ordered (C)

April 22, 2025 at 2:22 PM
This example is hard to understand from e.g. IGV/Ribbon (see Fig1) but pretty simple in SVTopo: 4 blocks deleted (B,D,F,H), 2 inverted (E,G), 1 re-ordered (C)
Reposted by Chris Saunders
I just released a new preprint! The manuscript describes SVTopo, a software tool that enhances visualization of complex SVs using HiFi data: www.biorxiv.org/content/10.1.... Here’s a summary of the results:

Complex structural variant visualization with SVTopo
Structural variants are genomic variants that impact at least 50 nucleotides and can play major roles in diversity and human health. Many structural variants are complex multi-breakpoint rearrangement...
www.biorxiv.org
April 22, 2025 at 2:21 PM
I just released a new preprint! The manuscript describes SVTopo, a software tool that enhances visualization of complex SVs using HiFi data: www.biorxiv.org/content/10.1.... Here’s a summary of the results:
Reposted by Chris Saunders

Last day to submit abstracts to #HiTSeq25 @hitseq.bsky.social #ISMB2025 - send us your work for an opportunity to present in the podium or poster.
Submit your abstract to HiTSeq at ISMB 2025 (July 20–24, Liverpool, UK)! Topics: short/long read analysis, assembly, variant detection, methylation, metagenomics, oncology, tools & more. Open to academia + industry. Deadline: April 17!
www.hitseq.org
www.hitseq.org
HiTSeq 2025: Conference on High Throughput Sequencing Algorithms and Applications - Home
www.hitseq.org
April 17, 2025 at 4:17 PM
Last day to submit abstracts to #HiTSeq25 @hitseq.bsky.social #ISMB2025 - send us your work for an opportunity to present in the podium or poster.
Reposted by Chris Saunders

Sawfish: Improving long-read structural variant discovery and genotyping with local haplotype modeling academic.oup.com/bioinformati... 🧬🖥️🧪 github.com/PacificBiosc...

April 11, 2025 at 3:30 PM
Sawfish: Improving long-read structural variant discovery and genotyping with local haplotype modeling academic.oup.com/bioinformati... 🧬🖥️🧪 github.com/PacificBiosc...
Reposted by Chris Saunders

Hello bluesky world! Newbee here! I have a postdoc position immediately available in my lab. It will focus on identifying high-quality transposons in many genomes and finding their impacts in evolution and traits. Most works, including EDTA2 development and annotation of 400+ genomes, are done! 1/n

April 10, 2025 at 6:08 PM
Hello bluesky world! Newbee here! I have a postdoc position immediately available in my lab. It will focus on identifying high-quality transposons in many genomes and finding their impacts in evolution and traits. Most works, including EDTA2 development and annotation of 400+ genomes, are done! 1/n
Reposted by Chris Saunders
🚀 Truvari v5.0 is here! 🎉
What’s new?
🔹 Enhanced symbolic variant support for, ,
🔹 Robust BND comparison for cross-representation SV matching
🔹 Improved SV sequence similarity & HUGE SV support
🔹 Cleaner UI & Revamped API
👉 More: github.com/ACEnglish/tr...
#Genomics #Bioinformatics
What’s new?
🔹 Enhanced symbolic variant support for
🔹 Robust BND comparison for cross-representation SV matching
🔹 Improved SV sequence similarity & HUGE SV support
🔹 Cleaner UI & Revamped API
👉 More: github.com/ACEnglish/tr...
#Genomics #Bioinformatics

GitHub - ACEnglish/truvari: Structural variant toolkit for VCFs
Structural variant toolkit for VCFs. Contribute to ACEnglish/truvari development by creating an account on GitHub.
github.com
January 10, 2025 at 1:43 PM
🚀 Truvari v5.0 is here! 🎉
What’s new?
🔹 Enhanced symbolic variant support for, ,
🔹 Robust BND comparison for cross-representation SV matching
🔹 Improved SV sequence similarity & HUGE SV support
🔹 Cleaner UI & Revamped API
👉 More: github.com/ACEnglish/tr...
#Genomics #Bioinformatics
What’s new?
🔹 Enhanced symbolic variant support for
🔹 Robust BND comparison for cross-representation SV matching
🔹 Improved SV sequence similarity & HUGE SV support
🔹 Cleaner UI & Revamped API
👉 More: github.com/ACEnglish/tr...
#Genomics #Bioinformatics
Great to see that sawfish, our new HiFi SV caller, is accepted for publication in Bioinformatics! Sawfish emphasizes local haplotype modeling to improve SV representation and genotyping in both single and joint-sample analysis. Advance-access article now available: (1/n)
doi.org/10.1093/bioi...
doi.org/10.1093/bioi...

Sawfish: Improving long-read structural variant discovery and genotyping with local haplotype modeling
AbstractMotivation. Structural variants (SVs) play an important role in evolutionary and functional genomics but are challenging to characterize. High-accu
doi.org
April 10, 2025 at 3:41 PM
Great to see that sawfish, our new HiFi SV caller, is accepted for publication in Bioinformatics! Sawfish emphasizes local haplotype modeling to improve SV representation and genotyping in both single and joint-sample analysis. Advance-access article now available: (1/n)
doi.org/10.1093/bioi...
doi.org/10.1093/bioi...
Reposted by Chris Saunders

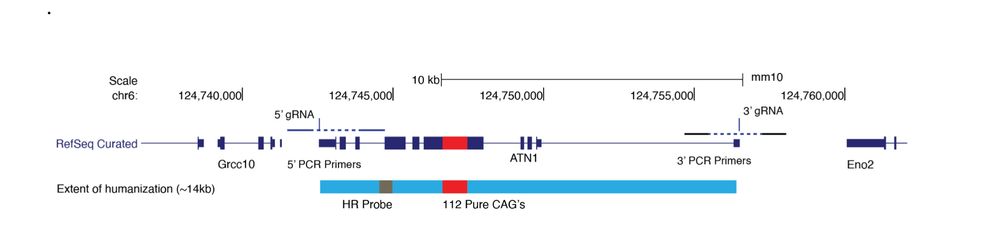
Very excited to see this study out in the world. We made what is (we think?) the first fully-humanized mouse model of a repeat expansion disorder, DRPLA, by replacing an entire copy of muring Atn1 with human ATN1 with 112 pure CAG repeats...
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
March 20, 2025 at 2:00 PM
Very excited to see this study out in the world. We made what is (we think?) the first fully-humanized mouse model of a repeat expansion disorder, DRPLA, by replacing an entire copy of muring Atn1 with human ATN1 with 112 pure CAG repeats...
www.biorxiv.org/content/10.1...
www.biorxiv.org/content/10.1...
Reposted by Chris Saunders
Xiao Chen #ACMGMtg25 describing Kivvi tool for assembling long repeat units in medically relevant genes (KIV2 and D4Z4) using #PacBio HiFi reads. Large repeats accurately assembled!

March 20, 2025 at 7:10 PM
Xiao Chen #ACMGMtg25 describing Kivvi tool for assembling long repeat units in medically relevant genes (KIV2 and D4Z4) using #PacBio HiFi reads. Large repeats accurately assembled!