Alan Amin
@alannawzadamin.bsky.social
180 followers
350 following
22 posts
Faculty fellow at NYU working with @andrewgwils.bsky.social. Statistics & machine learning for proteins, RNA, DNA.
Prev: @jura.bsky.social, PhD with Debora Marks
Website: alannawzadamin.github.io
Posts
Media
Videos
Starter Packs
Pinned








Alan Amin
@alannawzadamin.bsky.social
· Jun 16

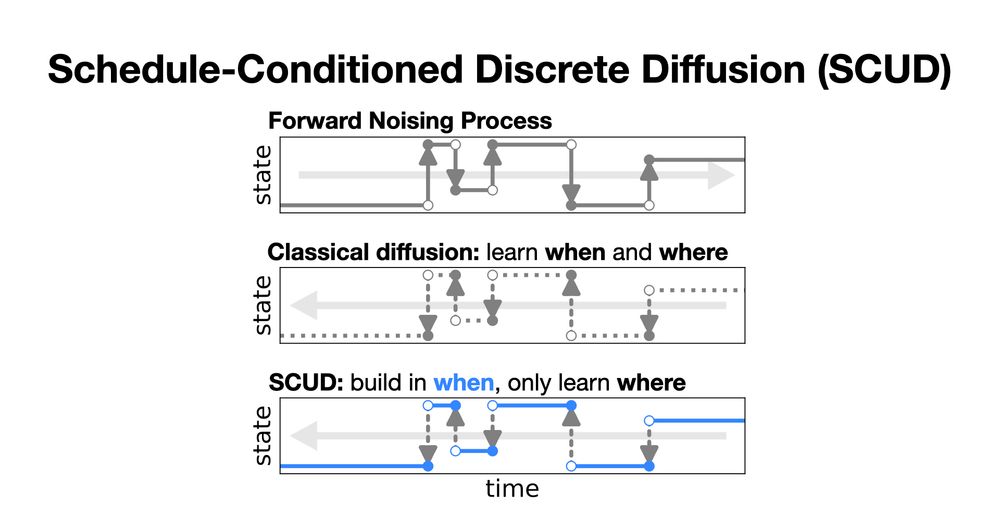
Why Masking Diffusion Works: Condition on the Jump Schedule for Improved Discrete Diffusion
Discrete diffusion models, like continuous diffusion models, generate high-quality samples by gradually undoing noise applied to datapoints with a Markov process. Gradual generation in theory comes wi...
arxiv.org





Reposted by Alan Amin

Eli Weinstein
@eliweinstein.bsky.social
· May 29
Reposted by Alan Amin

Jonathan Frazer
@jonnyfrazer.bsky.social
· May 26

Charlie Pugh
@cwjpugh.bsky.social
· May 26

From Likelihood to Fitness: Improving Variant Effect Prediction in Protein and Genome Language Models
Generative models trained on natural sequences are increasingly used to predict the effects of genetic variation, enabling progress in therapeutic design, disease risk prediction, and synthetic biolog...
www.biorxiv.org
Alan Amin
@alannawzadamin.bsky.social
· Dec 17

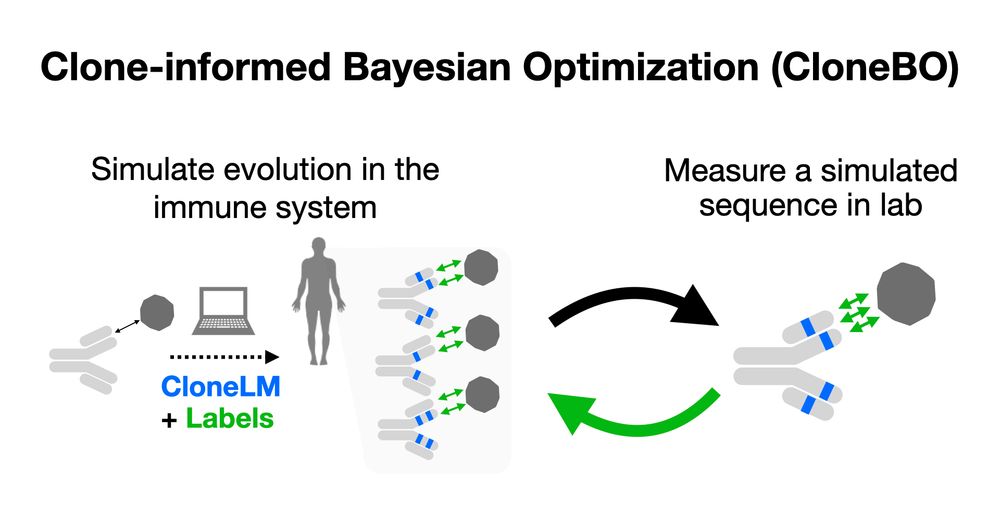
Bayesian Optimization of Antibodies Informed by a Generative Model of Evolving Sequences
To build effective therapeutics, biologists iteratively mutate antibody sequences to improve binding and stability. Proposed mutations can be informed by previous measurements or by learning from larg...
arxiv.org