
Posts
Media
Videos
Starter Packs
Reposted by Antonio Camargo



Antonio Camargo
@apcamargo.bsky.social
· Jul 17
Reposted by Antonio Camargo





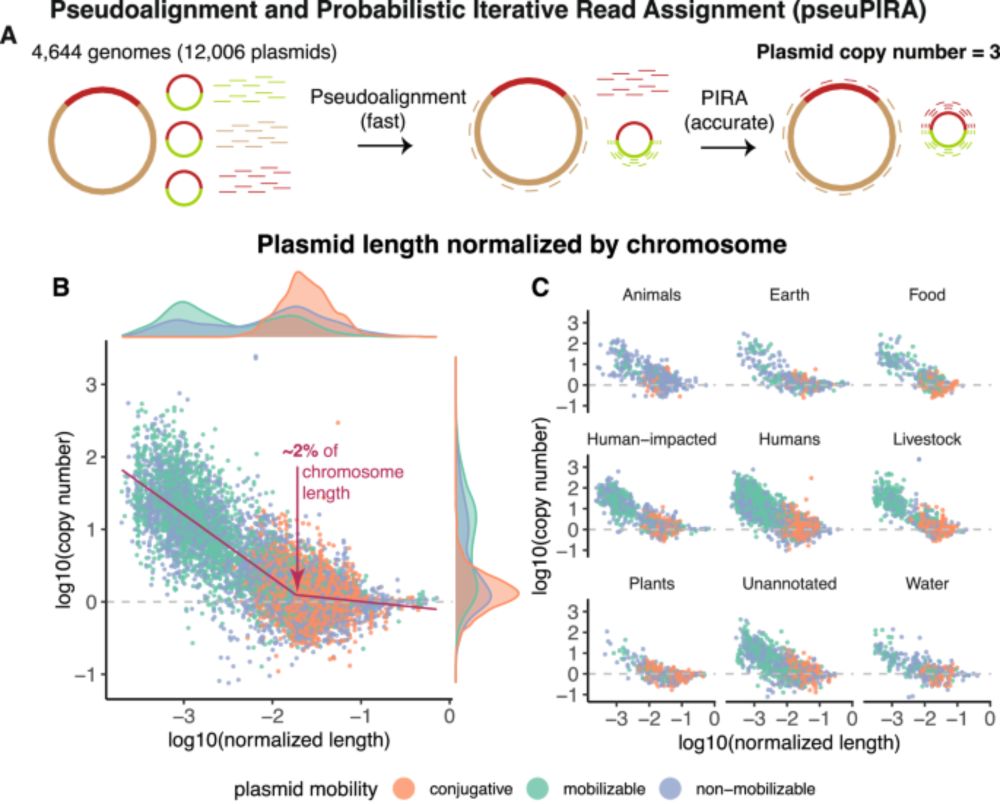
Antonio Camargo
@apcamargo.bsky.social
· Jun 25
Antonio Camargo
@apcamargo.bsky.social
· Jun 25
Antonio Camargo
@apcamargo.bsky.social
· Jun 21
Antonio Camargo
@apcamargo.bsky.social
· Jun 15
Reposted by Antonio Camargo

Reposted by Antonio Camargo
Reposted by Antonio Camargo




Reposted by Antonio Camargo

Reposted by Antonio Camargo


Reposted by Antonio Camargo

Eduardo Amorim
@cegamorim.bsky.social
· May 15
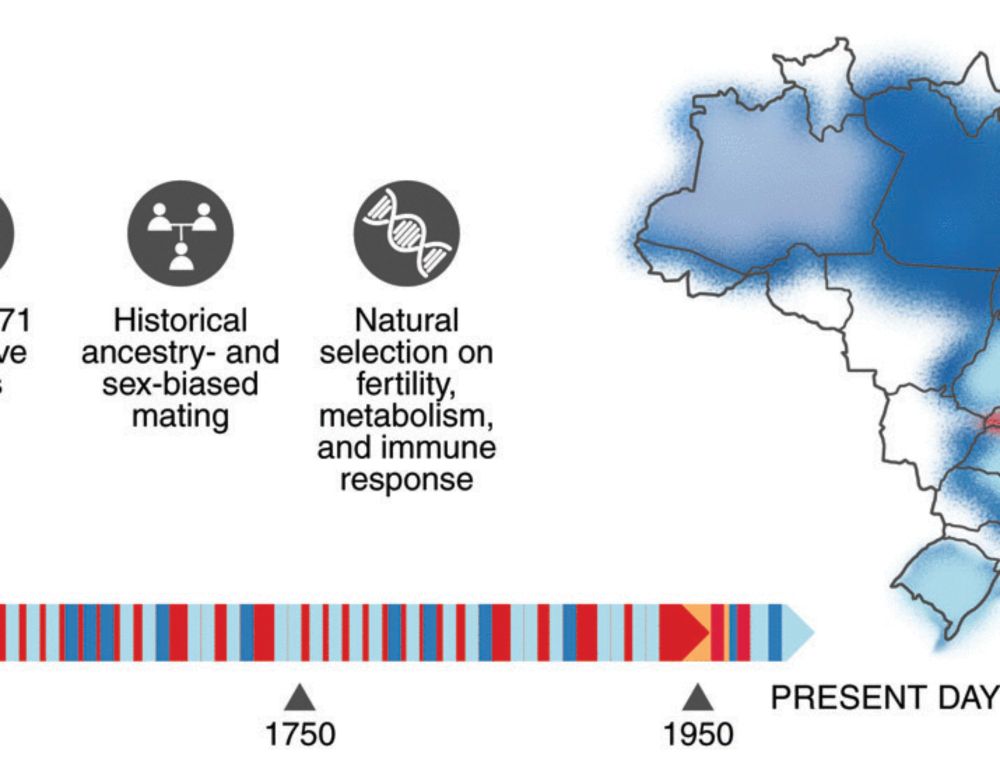
Admixture’s impact on Brazilian population evolution and health
Brazil, the largest Latin American country, is underrepresented in genomic research despite boasting the world’s largest recently admixed population. In this study, we generated 2723 high-coverage who...
www.science.org
Reposted by Antonio Camargo

Antonio Camargo
@apcamargo.bsky.social
· Apr 26

Clarification about rescued circular contigs · Issue #6 · GaetanBenoitDev/metaMDBG
Thanks for the work in MetaMDBG! In the README you mention that the _rc suffix flags "rescued circular " contigs and that the circularity of such sequences is not as reliable. However, I couldn't f...
github.com
Antonio Camargo
@apcamargo.bsky.social
· Apr 26
Reposted by Antonio Camargo


Antonio Camargo
@apcamargo.bsky.social
· Apr 26