
Peter Spackman
@crystalexplorer.net
370 followers
380 following
28 posts
Computers, crystals and chemistry. Research Fellow in the Computational Materials and Minerals Group at Curtin University. @[email protected]
Posts
Media
Videos
Starter Packs


Reposted by Peter Spackman

Volker Deringer
@vlderinger.bsky.social
· Aug 21

An automated framework for exploring and learning potential-energy surfaces - Nature Communications
Machine learning is revolutionising materials modelling but requires high-quality training data. Here, the authors introduce autoplex, an open framework automating exploration and fitting of potential...
doi.org


Reposted by Peter Spackman

Andrew S. Rosen
@andrewrosen.bsky.social
· Aug 14

An accurate and efficient framework for modelling the surface chemistry of ionic materials - Nature Chemistry
Simulating molecular adsorption on surfaces presents considerable challenges, as computational methods typically suffer from either insufficient accuracy or prohibitive computational costs. Now, with ...
www.nature.com
Reposted by Peter Spackman


Peter Spackman
@crystalexplorer.net
· Jul 30
Peter Spackman
@crystalexplorer.net
· Jul 30
Peter Spackman
@crystalexplorer.net
· Jul 29
Peter Spackman
@crystalexplorer.net
· Jul 29
Peter Spackman
@crystalexplorer.net
· Jul 29
Peter Spackman
@crystalexplorer.net
· Jul 9
Reposted by Peter Spackman


Reposted by Peter Spackman
Peter Spackman
@crystalexplorer.net
· May 20

Switching Perspectives on Hirshfeld Partitioning Schemes
We explore the foundations and applications of Hirshfeld partitioning, a fundamental technique used in the analysis of atoms in molecules, and the related Hirshfeld surface used to study molecules in ...
pubs.acs.org
Peter Spackman
@crystalexplorer.net
· May 15




Peter Spackman
@crystalexplorer.net
· Mar 5
Peter Spackman
@crystalexplorer.net
· Mar 5
Peter Spackman
@crystalexplorer.net
· Mar 5

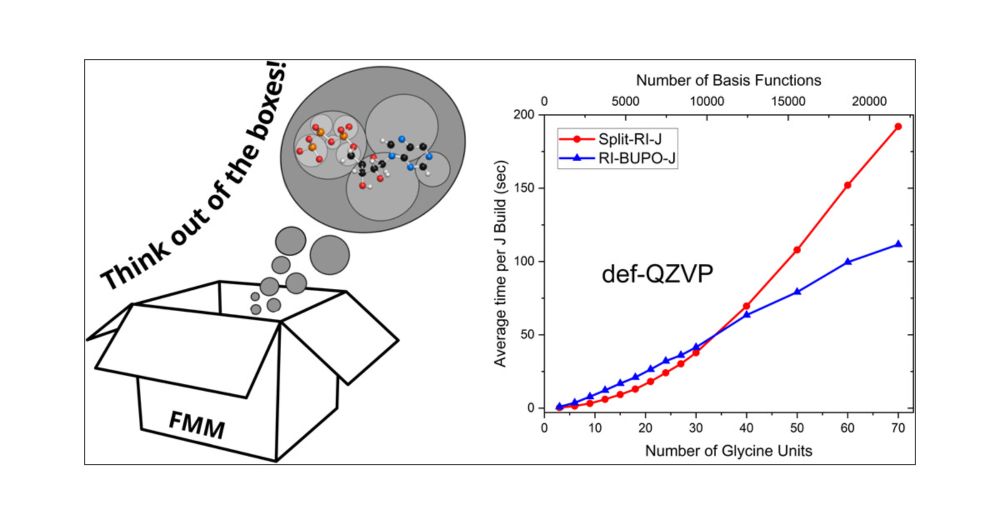
Reposted by Peter Spackman


Peter Spackman
@crystalexplorer.net
· Feb 15