Erin Young
@erinyoung.bsky.social
3.4K followers
2.3K following
320 posts
Public Health #Bioinformatician. Wants to sequence ALL THE THINGS. Personal account with alternative spellings and grammar structures. She/her
Posts
Media
Videos
Starter Packs



Erin Young
@erinyoung.bsky.social
· Sep 8
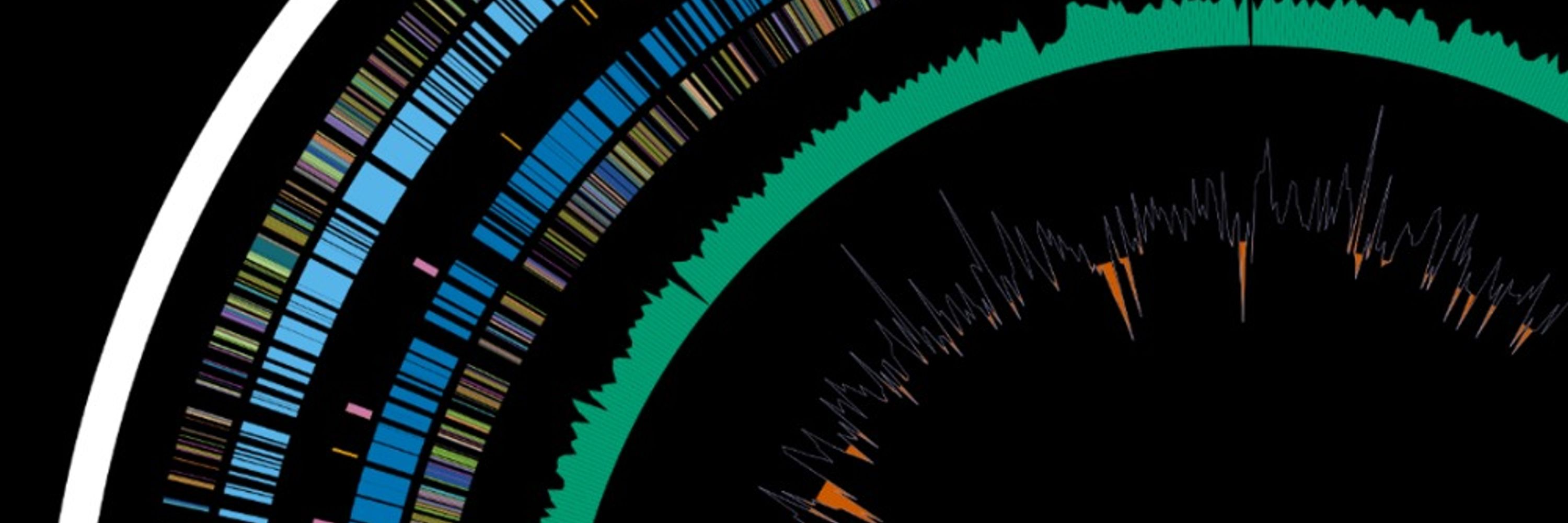
Mash Sketch of RefSeq Bacterial Reference Genomes
The mash reference that can be downloaded from the mash documentaion is for RefSeq version 70. I do not inherently have a problem with RefSeq version 70, but RefSeq is well past version 200 now. RefS...
zenodo.org
Erin Young
@erinyoung.bsky.social
· Aug 28
Erin Young
@erinyoung.bsky.social
· Aug 28
Erin Young
@erinyoung.bsky.social
· Aug 28
Erin Young
@erinyoung.bsky.social
· Aug 28
Erin Young
@erinyoung.bsky.social
· Aug 28
Erin Young
@erinyoung.bsky.social
· Aug 28

Erin Young
@erinyoung.bsky.social
· Aug 28
Reposted by Erin Young

Kate Baker
@ksbakes.bsky.social
· Aug 27

The WHO Bacterial Priority Pathogens List 2024: a prioritisation study to guide research, development, and public health strategies against antimicrobial resistance
The 2024 WHO BPPL is a key tool for prioritising research and development investments
and informing global public health policies to combat AMR. Gram-negative bacteria
and rifampicin-resistant M tuber...
www.thelancet.com
Erin Young
@erinyoung.bsky.social
· Aug 19
Erin Young
@erinyoung.bsky.social
· Aug 12

Erin Young
@erinyoung.bsky.social
· Aug 8
Erin Young
@erinyoung.bsky.social
· Aug 8