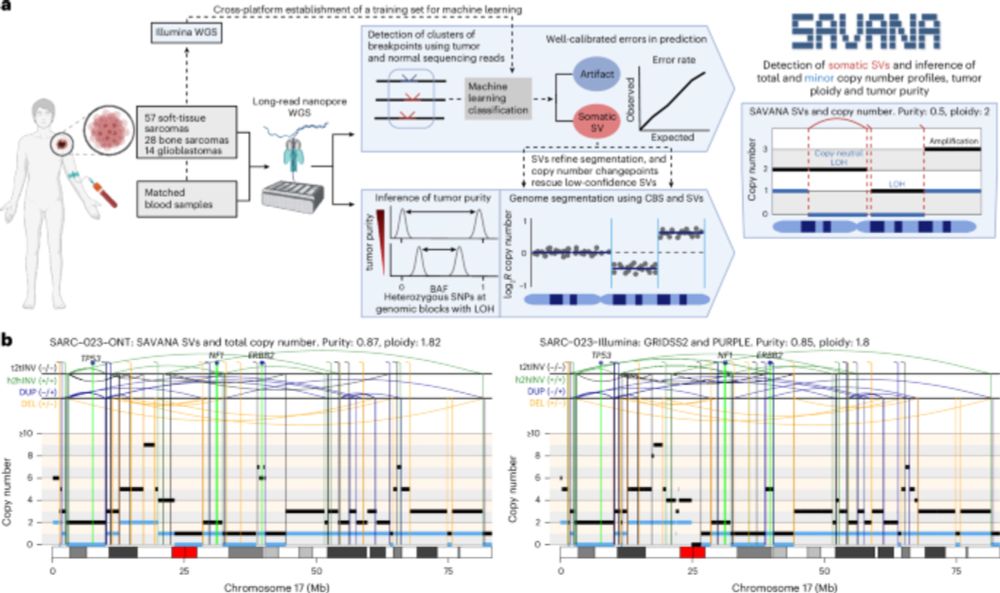
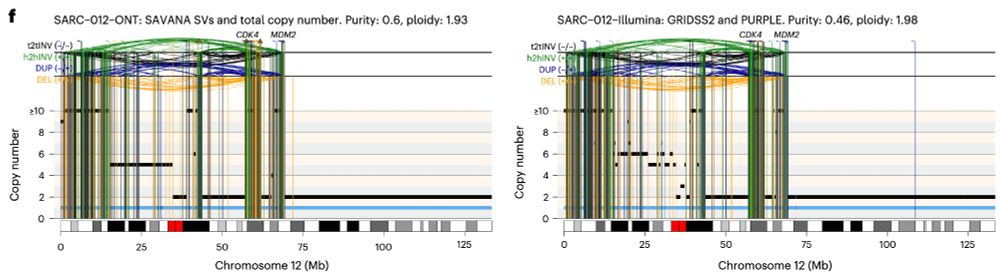
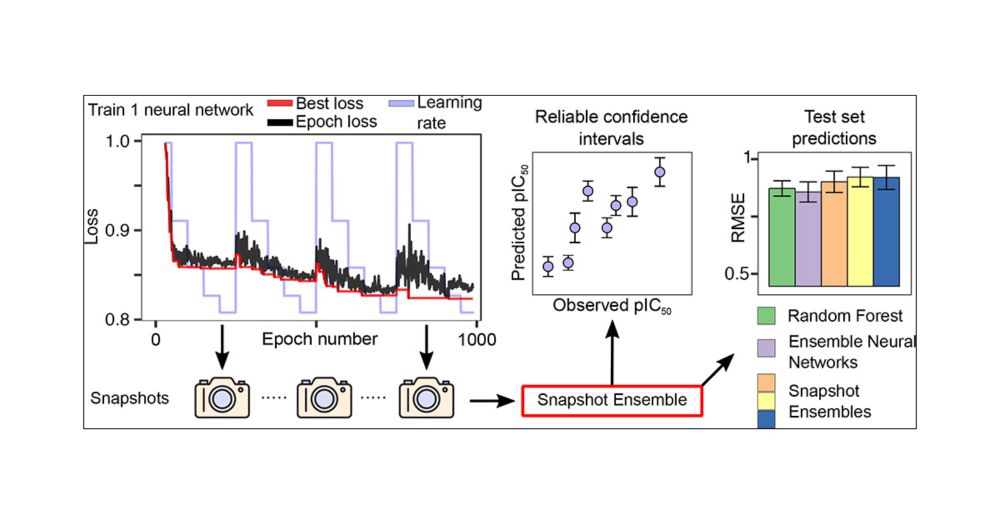
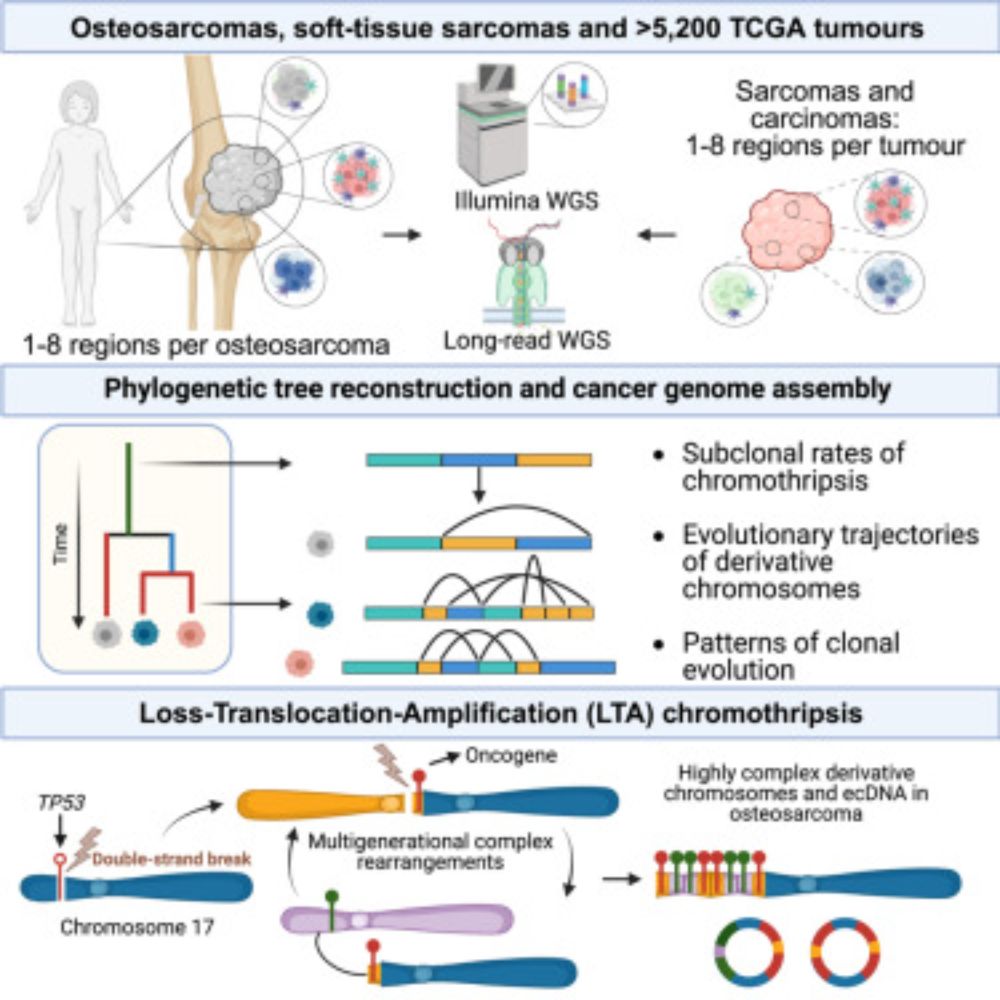
Posts
Media
Videos
Starter Packs


Reposted

EMBL-EBI
@ebi.embl.org
· May 29

Reposted