Bloom lab
@jbloomlab.bsky.social
14K followers
820 following
320 posts
Lab studying molecular evolution of proteins and viruses. Affiliated with Fred Hutch & HHMI.
https://jbloomlab.org/
Posts
Media
Videos
Starter Packs







Bloom lab
@jbloomlab.bsky.social
· Sep 4




Bloom lab
@jbloomlab.bsky.social
· Sep 4




Bloom lab
@jbloomlab.bsky.social
· Sep 4

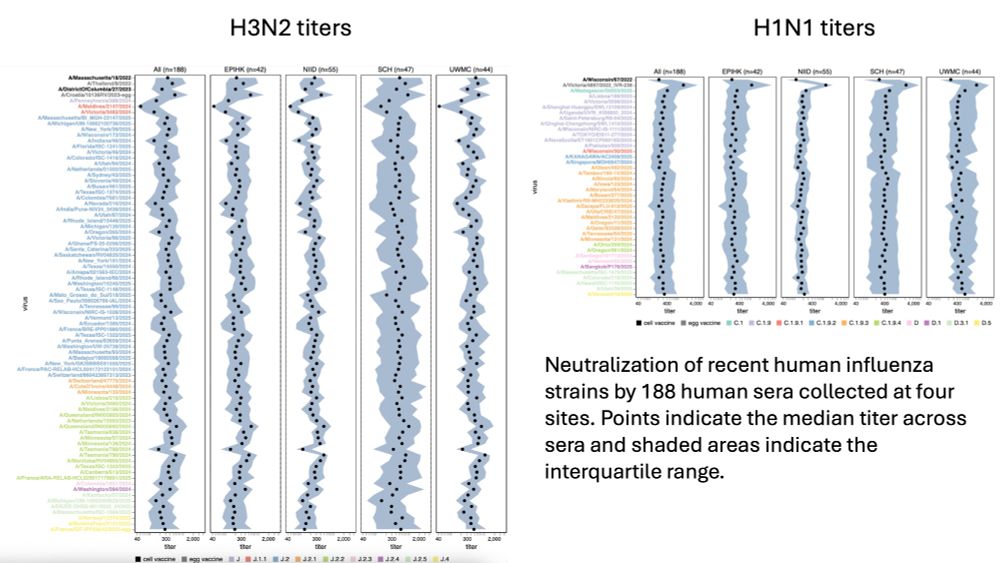
Determinants of human versus mosquito cell entry by the Chikungunya virus envelope proteins
Chikungunya virus (CHIKV) infects both humans and mosquitoes during its transmission cycle. How the virus’s envelope proteins mediate entry in cells from such different species is unclear. MXRA8 is a ...
www.biorxiv.org
