Studying cancer evolution needs multi-region or single cell seq for phylogenetics, right? Amazingly (I think!) we found single-sample bulk methylation suffices, via analysis of "fluctuating methylation". In @nature.com today led by brilliant @calumgabbutt.bsky.socialwww.nature.com/articles/s41...
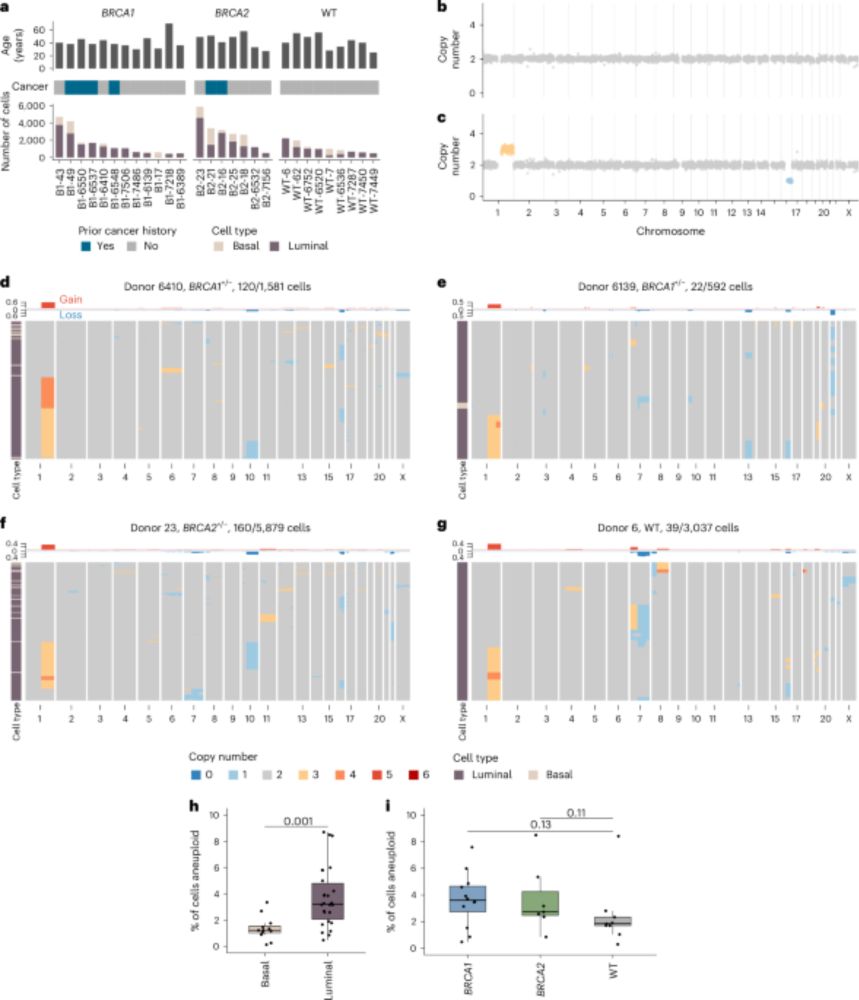
For the highly aneuploid cells that look like cancer genomes, I would guess they are some kind of pre-cancer clone and there may have been some kind of pre-malignant lesion that was too small for us to see in the pathology in most cases.
For the cells with just 1 or 2 of the common CNAs I think they likely provide a selective advantage to cells, or at least are not very deleterious relative to other CNAs.
Thanks to all co-authors, in particular @mike-oli.bsky.social, Vinci Au, Sam Aparicio, Joan Brugge and Sohrab Shah. This was a fascinating dataset to explore together!
We also found a very rare subset of cells that had extreme levels of aneuploidy, and to our eyes, were largely indistinguishable from breast cancer genomes.