



Max Fürst
@maxfus.bsky.social
Asst. Prof. Uni Groningen 🇳🇱
Comp & Exp Biochemist, Protein Engineer, 'Would-be designer' (F. Arnold) | SynBio | HT Screens & Selections | Nucleic Acid Enzymes | Biocatalysis | Rstats & Datavis
https://www.fuerstlab.com
https://orcid.org/0000-0001-7720-9
Comp & Exp Biochemist, Protein Engineer, 'Would-be designer' (F. Arnold) | SynBio | HT Screens & Selections | Nucleic Acid Enzymes | Biocatalysis | Rstats & Datavis
https://www.fuerstlab.com
https://orcid.org/0000-0001-7720-9
Interestingly, even though this demonstrates that the low designability of AFDB is essentially an artefact from RMSD calcs after alignment, using more sophisticated aligners (TMalgin, Sheba) do not achieve the same rescue.
16/19
16/19

December 16, 2025 at 3:32 PM
Interestingly, even though this demonstrates that the low designability of AFDB is essentially an artefact from RMSD calcs after alignment, using more sophisticated aligners (TMalgin, Sheba) do not achieve the same rescue.
16/19
16/19
Indeed, we show that if you truncate AFDB strucs to their corresponding PDB entry length, you get the same designability.
Without a PDB to compare to, you can also rescue via a simple trick: just compute statistical outliers from an initial alignment, realign without them, and use median RMSD
15/19
Without a PDB to compare to, you can also rescue via a simple trick: just compute statistical outliers from an initial alignment, realign without them, and use median RMSD
15/19

December 16, 2025 at 3:31 PM
Indeed, we show that if you truncate AFDB strucs to their corresponding PDB entry length, you get the same designability.
Without a PDB to compare to, you can also rescue via a simple trick: just compute statistical outliers from an initial alignment, realign without them, and use median RMSD
15/19
Without a PDB to compare to, you can also rescue via a simple trick: just compute statistical outliers from an initial alignment, realign without them, and use median RMSD
15/19
Realizing that this gap is RMSD, not pLDDT driven, we speculated the cause to be a PDB artefact: in xray strucs, flexible termini often lack density / unstructured parts were truncated to begin with. As a result, the designability of PDB structures gets very high compared to full-length AFDBs
14/19
14/19

December 16, 2025 at 3:30 PM
Realizing that this gap is RMSD, not pLDDT driven, we speculated the cause to be a PDB artefact: in xray strucs, flexible termini often lack density / unstructured parts were truncated to begin with. As a result, the designability of PDB structures gets very high compared to full-length AFDBs
14/19
14/19
We also question whether the refolding pipeline always is a robust evaluation metric to begin with. As e.g. noted in @moalquraishi.bsky.social's Genie2 paper, the designability of PDB structures is on average much higher than that of the AFDB. Now, we have a pretty good guess why:
13/19
13/19

December 16, 2025 at 3:29 PM
We also question whether the refolding pipeline always is a robust evaluation metric to begin with. As e.g. noted in @moalquraishi.bsky.social's Genie2 paper, the designability of PDB structures is on average much higher than that of the AFDB. Now, we have a pretty good guess why:
13/19
13/19
Sequences from design models like ProteinMPNN boost folding success, but for anything beyond medium sized proteins, you almost never can accurately fold in ss mode.
11/19
11/19

December 16, 2025 at 3:27 PM
Sequences from design models like ProteinMPNN boost folding success, but for anything beyond medium sized proteins, you almost never can accurately fold in ss mode.
11/19
11/19
We next looked at a set of literature-reported experimentally tested designs and compared folding models’ ability to act as “oracles”. Again, evo info was detrimental. MSA mode’s poor performance can be obfuscated though, for seqs where the MSA is very shallow / empty.
Duh: empty MSA == ss mode
9/19
Duh: empty MSA == ss mode
9/19

December 16, 2025 at 3:25 PM
We next looked at a set of literature-reported experimentally tested designs and compared folding models’ ability to act as “oracles”. Again, evo info was detrimental. MSA mode’s poor performance can be obfuscated though, for seqs where the MSA is very shallow / empty.
Duh: empty MSA == ss mode
9/19
Duh: empty MSA == ss mode
9/19
Worse: If you repeat this experiment starting from sequences designed with ProteinMPNN, the effect is even worse. The reason likely is that ProteinMPNN designs very strongly and unambiguously encode the intended structure, which can make folding models overconfident about their predictions.
8/19
8/19

December 16, 2025 at 3:24 PM
Worse: If you repeat this experiment starting from sequences designed with ProteinMPNN, the effect is even worse. The reason likely is that ProteinMPNN designs very strongly and unambiguously encode the intended structure, which can make folding models overconfident about their predictions.
8/19
8/19
We found that this trend is exacerbated by the availability of evolutionary info to models: AF2 MSA behaves the worst, ESMfold is a bit better, and AF ss is the best (out of bad bunch).
As others noted: the signal from these MSAs / pLM embeddings overrules “reason”
7/19
As others noted: the signal from these MSAs / pLM embeddings overrules “reason”
7/19

December 16, 2025 at 3:23 PM
We found that this trend is exacerbated by the availability of evolutionary info to models: AF2 MSA behaves the worst, ESMfold is a bit better, and AF ss is the best (out of bad bunch).
As others noted: the signal from these MSAs / pLM embeddings overrules “reason”
7/19
As others noted: the signal from these MSAs / pLM embeddings overrules “reason”
7/19
We probed this for ESMfold, and for AF2, where two modes can be employed: the default MSA mode (often seen for redesign of native proteins) and single sequence mode (often seen for de novo protein design)
First, we checked how good they are at identifying clearly bad designs
5/19
First, we checked how good they are at identifying clearly bad designs
5/19

December 16, 2025 at 3:21 PM
We probed this for ESMfold, and for AF2, where two modes can be employed: the default MSA mode (often seen for redesign of native proteins) and single sequence mode (often seen for de novo protein design)
First, we checked how good they are at identifying clearly bad designs
5/19
First, we checked how good they are at identifying clearly bad designs
5/19
Led by @kerlenkorbeld.bsky.social and Seva Viliuga, we started this project under the premise of the field's most common protein design evaluation workflow: the self-consistency pipeline (function->backbone->sequence->evaluation), where folding models are used for the last step.
2/19
2/19

December 16, 2025 at 3:20 PM
Led by @kerlenkorbeld.bsky.social and Seva Viliuga, we started this project under the premise of the field's most common protein design evaluation workflow: the self-consistency pipeline (function->backbone->sequence->evaluation), where folding models are used for the last step.
2/19
2/19
New preprint🚨
Imagine (re)designing a protein via inverse folding. AF2 predicts the designed sequence to a structure with pLDDT 94 & you get 1.8 Å RMSD to the input. Perfect design?
What if I told u that the structure has 4 solvent-exposed Trp and 3 Pro where a Gly should be?
Why to be wary🧵👇
Imagine (re)designing a protein via inverse folding. AF2 predicts the designed sequence to a structure with pLDDT 94 & you get 1.8 Å RMSD to the input. Perfect design?
What if I told u that the structure has 4 solvent-exposed Trp and 3 Pro where a Gly should be?
Why to be wary🧵👇
December 16, 2025 at 3:15 PM
New preprint🚨
Imagine (re)designing a protein via inverse folding. AF2 predicts the designed sequence to a structure with pLDDT 94 & you get 1.8 Å RMSD to the input. Perfect design?
What if I told u that the structure has 4 solvent-exposed Trp and 3 Pro where a Gly should be?
Why to be wary🧵👇
Imagine (re)designing a protein via inverse folding. AF2 predicts the designed sequence to a structure with pLDDT 94 & you get 1.8 Å RMSD to the input. Perfect design?
What if I told u that the structure has 4 solvent-exposed Trp and 3 Pro where a Gly should be?
Why to be wary🧵👇
I set myself a reminder to check this 1 year later & turns out I was mostly wrong🥲
Here search results from on "DNA", last year (orange) vs now (blue) and the indexing issue is only noticeable for the current year ('24), barely an increase for '23
New hypothesis: all (bio?) topics peaked around '21?
Here search results from on "DNA", last year (orange) vs now (blue) and the indexing issue is only noticeable for the current year ('24), barely an increase for '23
New hypothesis: all (bio?) topics peaked around '21?

November 5, 2025 at 4:06 PM
I set myself a reminder to check this 1 year later & turns out I was mostly wrong🥲
Here search results from on "DNA", last year (orange) vs now (blue) and the indexing issue is only noticeable for the current year ('24), barely an increase for '23
New hypothesis: all (bio?) topics peaked around '21?
Here search results from on "DNA", last year (orange) vs now (blue) and the indexing issue is only noticeable for the current year ('24), barely an increase for '23
New hypothesis: all (bio?) topics peaked around '21?
wouldn't it make much more sense to plot this relative to normal distribution found across the proteome?
Too lazy to extract the numbers and make a scatter plot, but I think Ala and Glu are at least as off the axis as the aromatics
www.uniprot.org/uniprotkb/st...
Too lazy to extract the numbers and make a scatter plot, but I think Ala and Glu are at least as off the axis as the aromatics
www.uniprot.org/uniprotkb/st...

October 27, 2025 at 5:14 PM
wouldn't it make much more sense to plot this relative to normal distribution found across the proteome?
Too lazy to extract the numbers and make a scatter plot, but I think Ala and Glu are at least as off the axis as the aromatics
www.uniprot.org/uniprotkb/st...
Too lazy to extract the numbers and make a scatter plot, but I think Ala and Glu are at least as off the axis as the aromatics
www.uniprot.org/uniprotkb/st...
Un-frigging-believable. I have waited for something like this for so long!
October 5, 2025 at 8:01 AM
Un-frigging-believable. I have waited for something like this for so long!
I kept telling people this in discussions, until someone German insisted so I looked it up and it's indeed not that clear.
Here formatted in a similar way from source www.bka.de/DE/AktuelleI...
Here formatted in a similar way from source www.bka.de/DE/AktuelleI...

August 8, 2025 at 9:43 PM
I kept telling people this in discussions, until someone German insisted so I looked it up and it's indeed not that clear.
Here formatted in a similar way from source www.bka.de/DE/AktuelleI...
Here formatted in a similar way from source www.bka.de/DE/AktuelleI...
Now that OpenCRISPR is in nature and rekindled the 'what's-a-novel-sequence' debate, I'm happy to share an app to check this, which I built for fun some time ago.
fuerstlab.shinyapps.io/SeqNovelty/
quick 🧵
fuerstlab.shinyapps.io/SeqNovelty/
quick 🧵
August 1, 2025 at 11:50 AM
Now that OpenCRISPR is in nature and rekindled the 'what's-a-novel-sequence' debate, I'm happy to share an app to check this, which I built for fun some time ago.
fuerstlab.shinyapps.io/SeqNovelty/
quick 🧵
fuerstlab.shinyapps.io/SeqNovelty/
quick 🧵
Fixed that for you!
Original (very awkward) color scale did not reflect how much of an outlier NL is.
Picture changes for 3yr - school age though.
Of course this data does not distinguish between day care vs grandparents etc. Not that the latter is an option for expats, anyway 😶🌫️
Original (very awkward) color scale did not reflect how much of an outlier NL is.
Picture changes for 3yr - school age though.
Of course this data does not distinguish between day care vs grandparents etc. Not that the latter is an option for expats, anyway 😶🌫️


June 2, 2025 at 8:16 AM
Fixed that for you!
Original (very awkward) color scale did not reflect how much of an outlier NL is.
Picture changes for 3yr - school age though.
Of course this data does not distinguish between day care vs grandparents etc. Not that the latter is an option for expats, anyway 😶🌫️
Original (very awkward) color scale did not reflect how much of an outlier NL is.
Picture changes for 3yr - school age though.
Of course this data does not distinguish between day care vs grandparents etc. Not that the latter is an option for expats, anyway 😶🌫️
As a final application example, we demonstrate protein immobiliziation. Commercial azido agarose reacted happily with ADD-tagged GFP, and we got a brightly shining resin after reacting the protein on-column with ApbE!

May 20, 2025 at 12:30 PM
As a final application example, we demonstrate protein immobiliziation. Commercial azido agarose reacted happily with ADD-tagged GFP, and we got a brightly shining resin after reacting the protein on-column with ApbE!
Conjugating commercial click-modified DNA oligos also worked at the first shot without optimization, although at relatively low conjugation yields. We’re not so experienced with the chemistry, but apparently Cu catalysis isn’t great for DNA either, so probably switching to SPAAC would work better.

May 20, 2025 at 12:30 PM
Conjugating commercial click-modified DNA oligos also worked at the first shot without optimization, although at relatively low conjugation yields. We’re not so experienced with the chemistry, but apparently Cu catalysis isn’t great for DNA either, so probably switching to SPAAC would work better.
As expected, biomolecule conjugation needs more care: we saw moderate conversion yields in protein-protein conjugations (N-N/C-C/branched). We did not optimize our proof of concept—safe to assume that it could easily be pushed by varying position, adding linkers, optimized reaction conditions, etc.

May 20, 2025 at 12:30 PM
As expected, biomolecule conjugation needs more care: we saw moderate conversion yields in protein-protein conjugations (N-N/C-C/branched). We did not optimize our proof of concept—safe to assume that it could easily be pushed by varying position, adding linkers, optimized reaction conditions, etc.
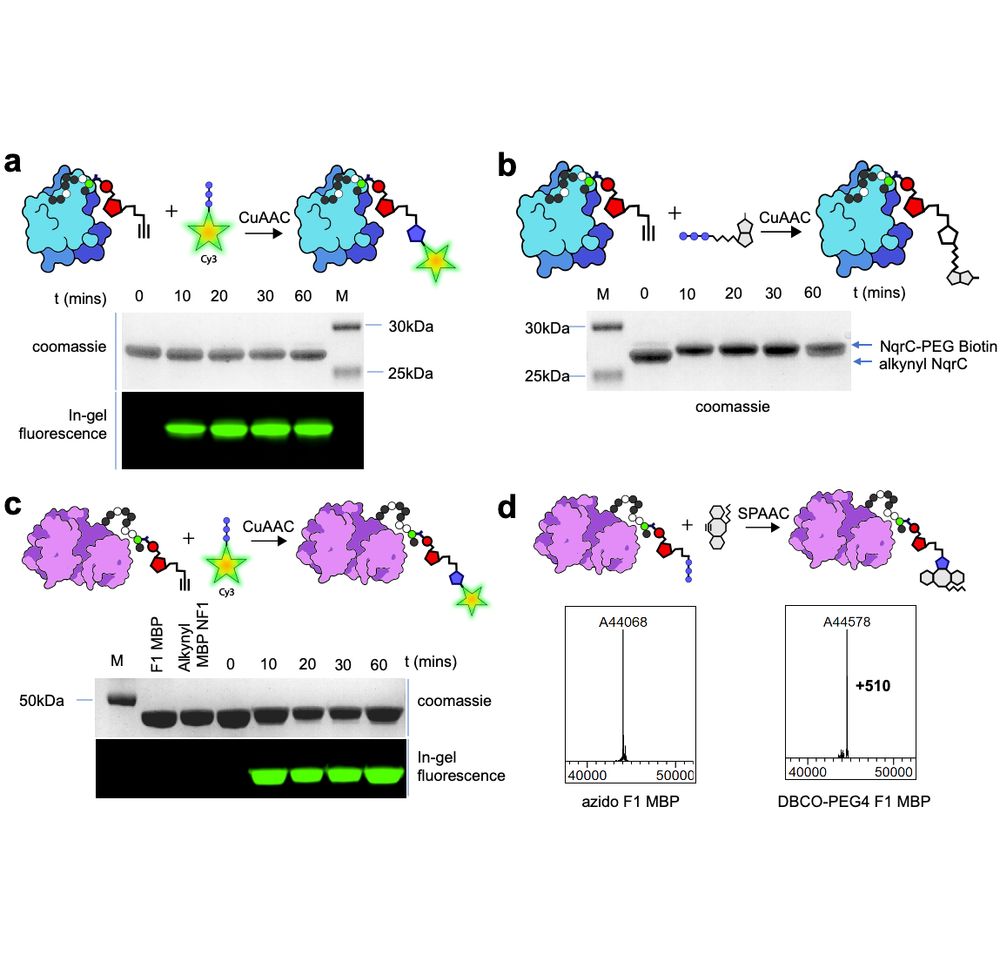
Small molecules were readily conjugated to all our example proteins: we show attachment of fluorophores & biotin, and establish that both classic copper-catalyzed, as well as @carolynbertozzi.bskyverified.social's copper-free SPAAC reaction are compatible with ADD tags & result in full labeling.
May 20, 2025 at 12:30 PM
Small molecules were readily conjugated to all our example proteins: we show attachment of fluorophores & biotin, and establish that both classic copper-catalyzed, as well as @carolynbertozzi.bskyverified.social's copper-free SPAAC reaction are compatible with ADD tags & result in full labeling.
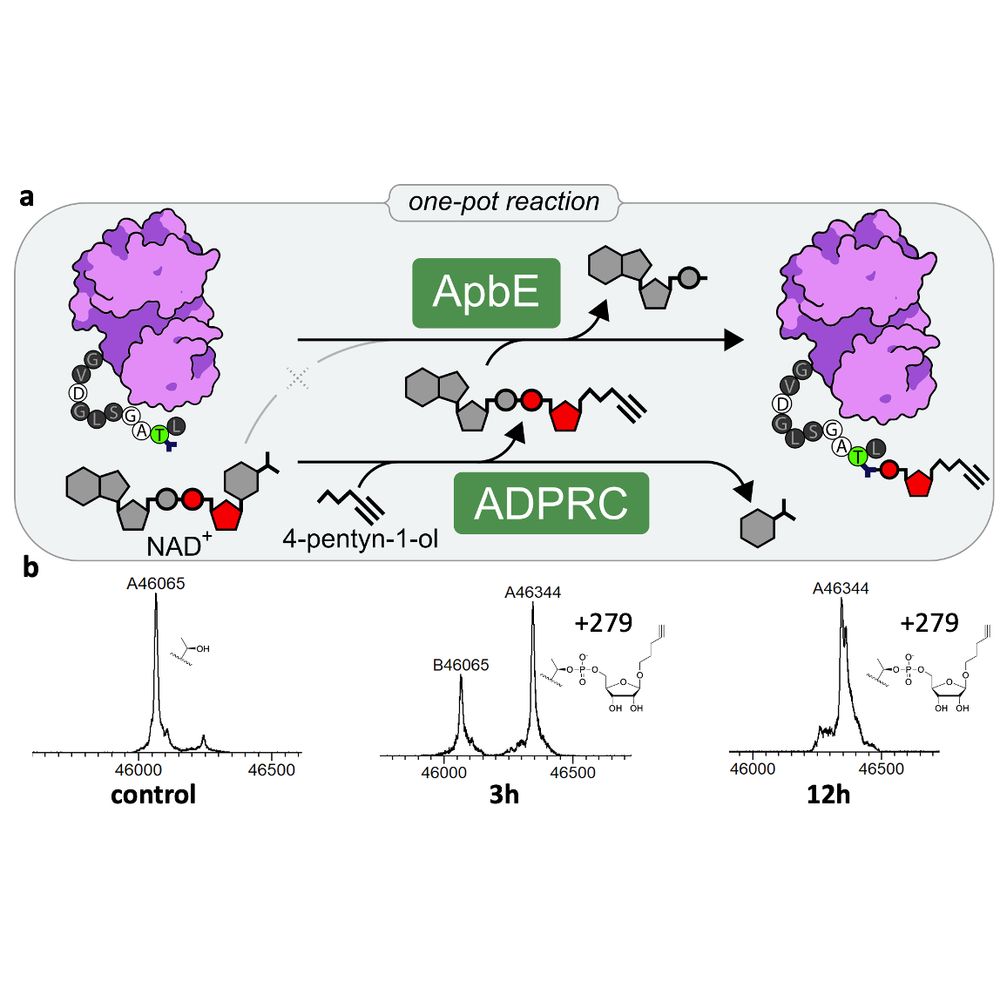
Because ADPRC works on NAD+, not NADPH, while ApbE only accepts NADPH and ignores NAD+, we can even run the two enzymatic steps (substrate generation & protein modification) in one pot—just add the two enzymes with the precursors NAD+ and the click alkanol to your target protein!
May 20, 2025 at 12:30 PM
Because ADPRC works on NAD+, not NADPH, while ApbE only accepts NADPH and ignores NAD+, we can even run the two enzymatic steps (substrate generation & protein modification) in one pot—just add the two enzymes with the precursors NAD+ and the click alkanol to your target protein!
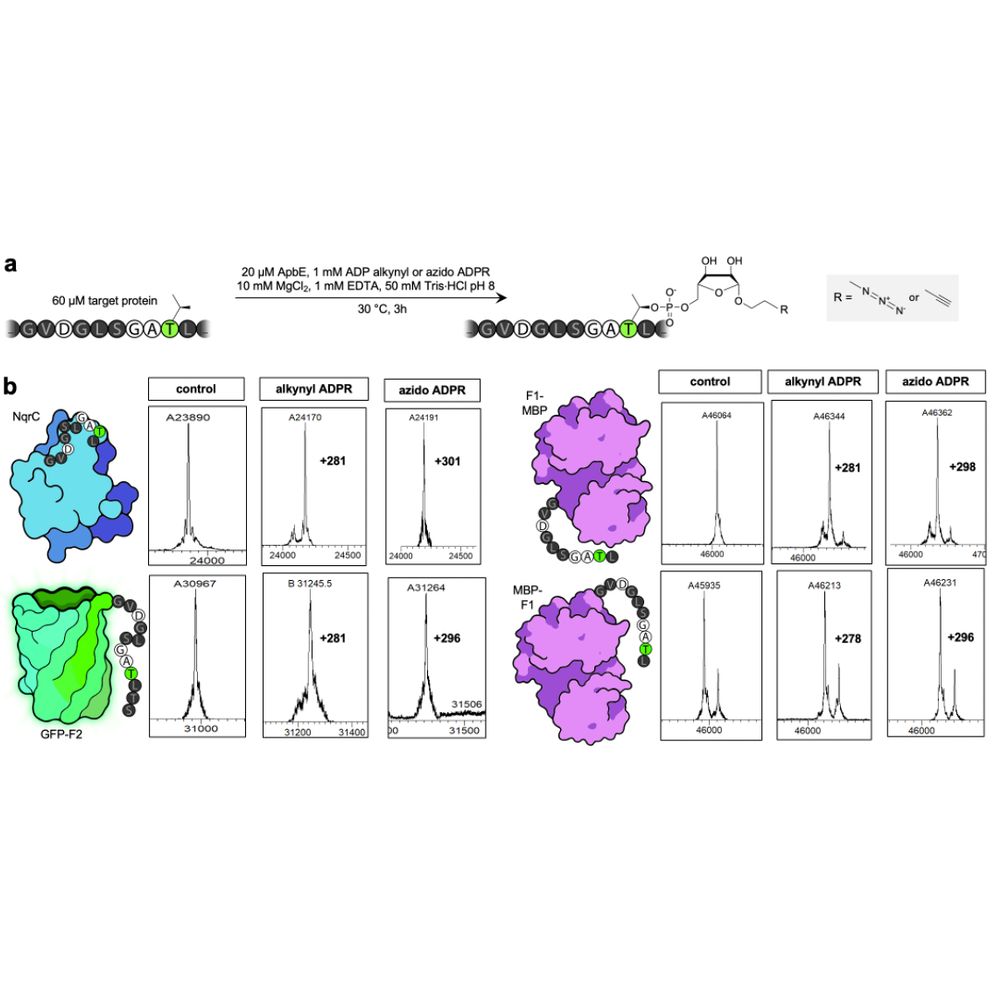
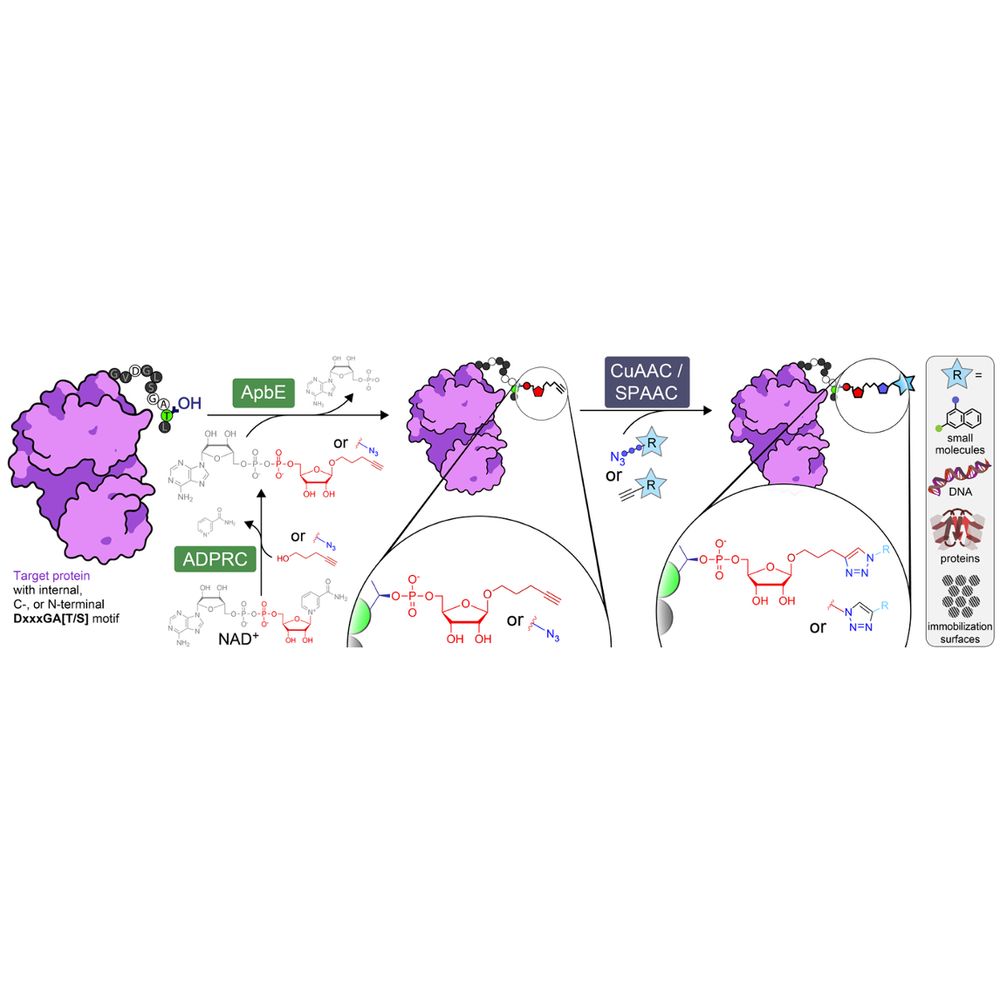
Turns out ApbE is happily accepting this substrate, allowing us to label a variety of proteins at N or C terminus, as well as internally, with either alkynyl or azido ribose phosphate moieties, yielding typically full conversion after a few hours of reaction, as determined by MS.
May 20, 2025 at 12:30 PM
Turns out ApbE is happily accepting this substrate, allowing us to label a variety of proteins at N or C terminus, as well as internally, with either alkynyl or azido ribose phosphate moieties, yielding typically full conversion after a few hours of reaction, as determined by MS.
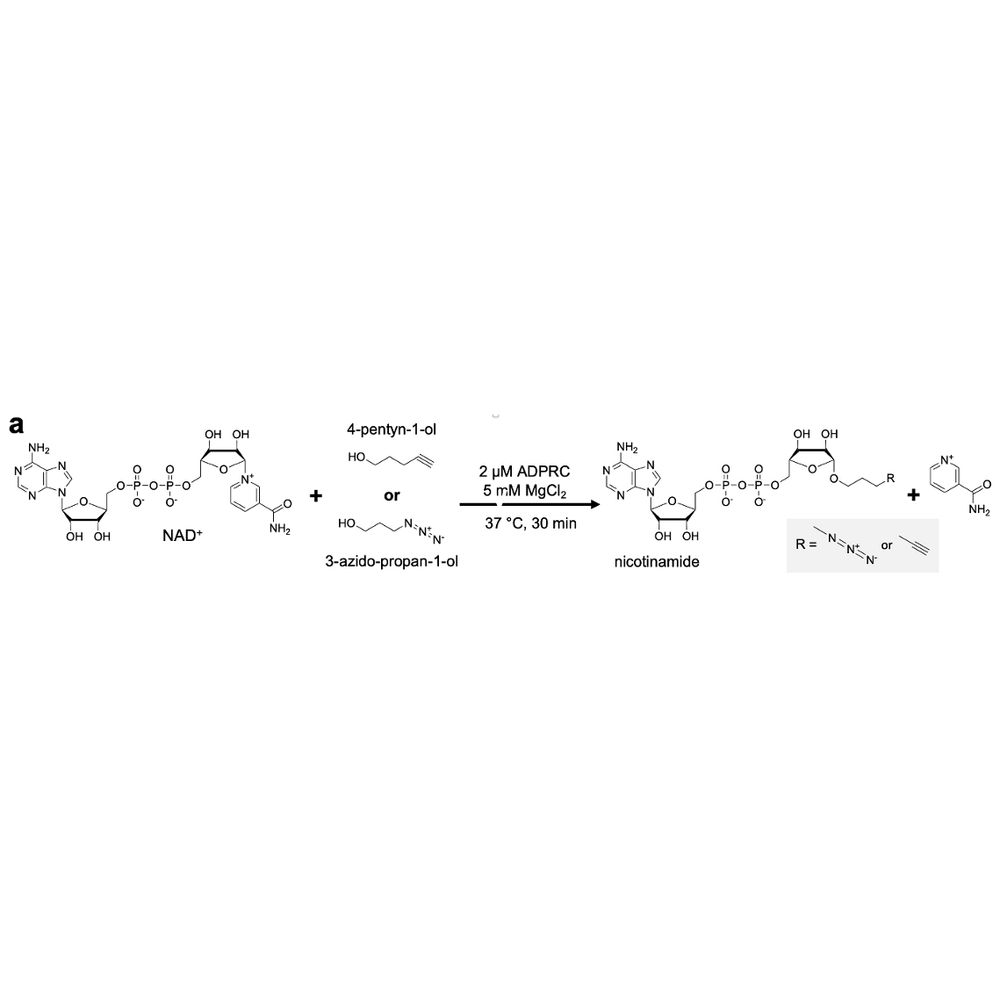
Given the enzyme’s reported promiscuity toward NADH, we hypothesised that it tolerates synthetic substrates with similar structure. Courtesy to Jäschke lab developing NAD capture seq, we knew that a commercial enzyme—ADPRC—can generate a click dinucleotide from cheap small molecule alcohols & NAD+
May 20, 2025 at 12:30 PM
Given the enzyme’s reported promiscuity toward NADH, we hypothesised that it tolerates synthetic substrates with similar structure. Courtesy to Jäschke lab developing NAD capture seq, we knew that a commercial enzyme—ADPRC—can generate a click dinucleotide from cheap small molecule alcohols & NAD+
🚨 preprint 2️⃣ this month: our (purely experimental🧪) venture into #ChemBio
We prouldy present: ADD-tagging of proteins (or "ADDing") —a super convenient enzymatic technique to install click chemistry handles on proteins.
Led by superstar @wahyuwidodo.bsky.social
www.biorxiv.org/content/10.1...
A 🧵👇🏽
We prouldy present: ADD-tagging of proteins (or "ADDing") —a super convenient enzymatic technique to install click chemistry handles on proteins.
Led by superstar @wahyuwidodo.bsky.social
www.biorxiv.org/content/10.1...
A 🧵👇🏽
May 20, 2025 at 12:30 PM
🚨 preprint 2️⃣ this month: our (purely experimental🧪) venture into #ChemBio
We prouldy present: ADD-tagging of proteins (or "ADDing") —a super convenient enzymatic technique to install click chemistry handles on proteins.
Led by superstar @wahyuwidodo.bsky.social
www.biorxiv.org/content/10.1...
A 🧵👇🏽
We prouldy present: ADD-tagging of proteins (or "ADDing") —a super convenient enzymatic technique to install click chemistry handles on proteins.
Led by superstar @wahyuwidodo.bsky.social
www.biorxiv.org/content/10.1...
A 🧵👇🏽