


Jo Ma
@myzzzz.bsky.social
I believe innovation sparks from the intersection of difference
-Revolutionizing Drug & Materials Design for Everyone with Computational Chemistry https://quantabricks.substack.com/p/molecular-docking-in-a-easy-way
-Revolutionizing Drug & Materials Design for Everyone with Computational Chemistry https://quantabricks.substack.com/p/molecular-docking-in-a-easy-way
I guess, HAT and PCET reactions records exist in their traning set. We do have some H-transfer benchmarking reaction path
June 13, 2025 at 1:56 AM
I guess, HAT and PCET reactions records exist in their traning set. We do have some H-transfer benchmarking reaction path
What surprised me is that UMA also handles extreme cases like CNT breaking. I am testing high-temperature combustion. So it's not just about small distortions—UMA might actually work as a super ReaxFF, we hope so.
June 13, 2025 at 1:55 AM
What surprised me is that UMA also handles extreme cases like CNT breaking. I am testing high-temperature combustion. So it's not just about small distortions—UMA might actually work as a super ReaxFF, we hope so.
In Section 2.4.2 of the OMol25 paper, the Interpolated Reactivity Datasets include transition states and reaction steps, which already go beyond equilibrium.
June 13, 2025 at 1:55 AM
In Section 2.4.2 of the OMol25 paper, the Interpolated Reactivity Datasets include transition states and reaction steps, which already go beyond equilibrium.
Our platform enables users to design molecules without requiring computational chemistry expertise, allowing them to focus on their scientific challenges.
If any drug discovery teams are interested, feel free to reach out.
If any drug discovery teams are interested, feel free to reach out.
March 21, 2025 at 9:28 PM
Our platform enables users to design molecules without requiring computational chemistry expertise, allowing them to focus on their scientific challenges.
If any drug discovery teams are interested, feel free to reach out.
If any drug discovery teams are interested, feel free to reach out.
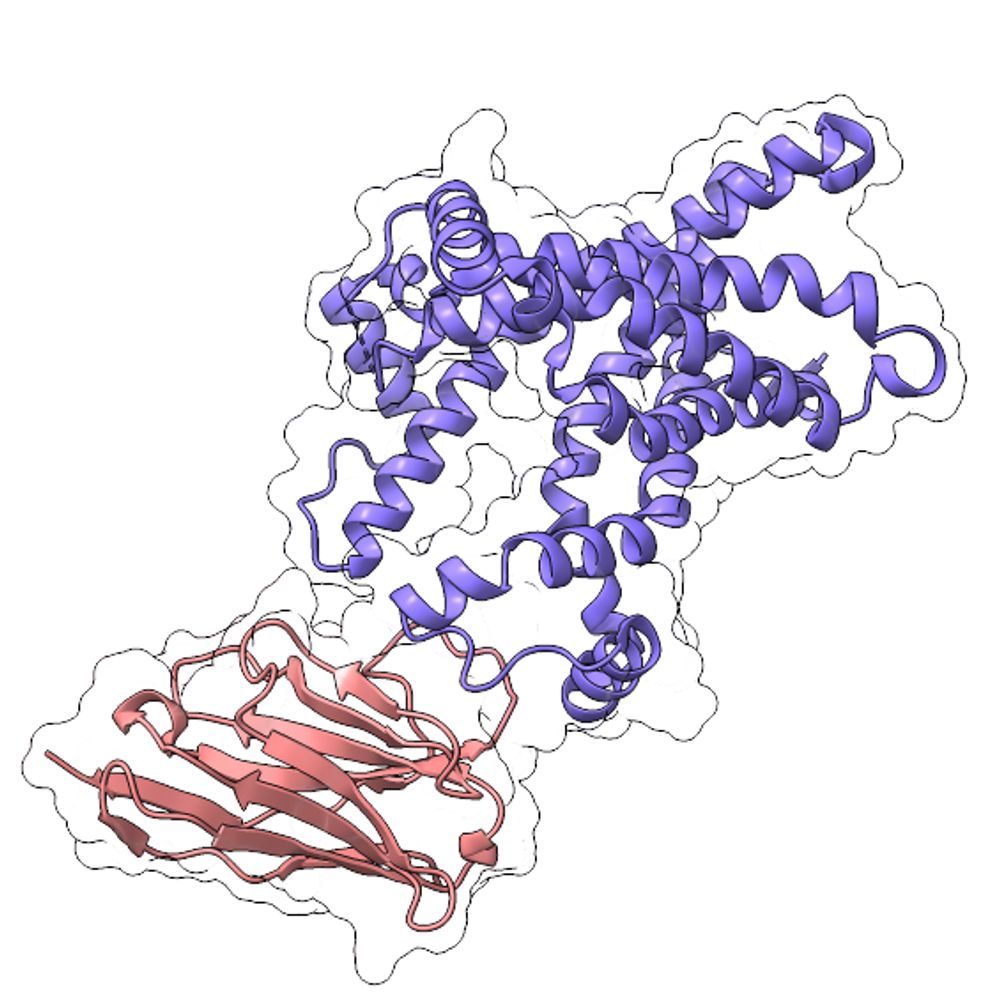
By pre-analyzing and identifying the docking pocket, I used the ChemOrchestra platform to optimize and dock 195 drug-like molecules efficiently. And I do it on pure CPU, so no huge computational cost.
March 21, 2025 at 6:05 PM
By pre-analyzing and identifying the docking pocket, I used the ChemOrchestra platform to optimize and dock 195 drug-like molecules efficiently. And I do it on pure CPU, so no huge computational cost.