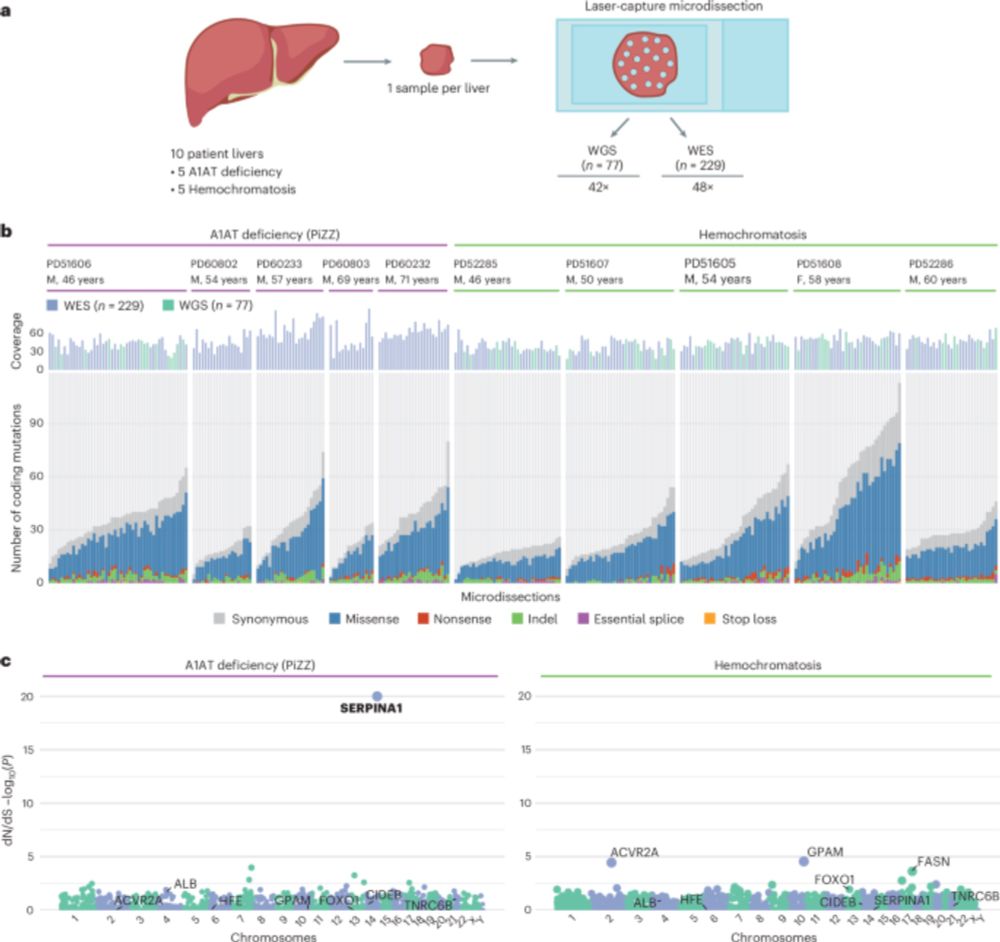
Natalia Brzozowska
@nataliabrzozowska.bsky.social
40 followers
68 following
13 posts
Researching somatic evolution in liver disease at Wellcome Sanger Institute
Posts
Media
Videos
Starter Packs