Ryan Wick
@rrwick.bsky.social
1.1K followers
120 following
78 posts
Bioinformatician at the Centre for Pathogen Genomics at the University of Melbourne
Posts
Media
Videos
Starter Packs


Ryan Wick
@rrwick.bsky.social
· 14d
Ryan Wick
@rrwick.bsky.social
· 14d
Ryan Wick
@rrwick.bsky.social
· 14d
Ryan Wick
@rrwick.bsky.social
· 15d



Reposted by Ryan Wick


Ryan Wick
@rrwick.bsky.social
· Sep 7
Ryan Wick
@rrwick.bsky.social
· Sep 7
Ryan Wick
@rrwick.bsky.social
· Sep 5
Ryan Wick
@rrwick.bsky.social
· Sep 5
Ryan Wick
@rrwick.bsky.social
· Sep 5
Ryan Wick
@rrwick.bsky.social
· Sep 5
Ryan Wick
@rrwick.bsky.social
· Sep 5
Reposted by Ryan Wick

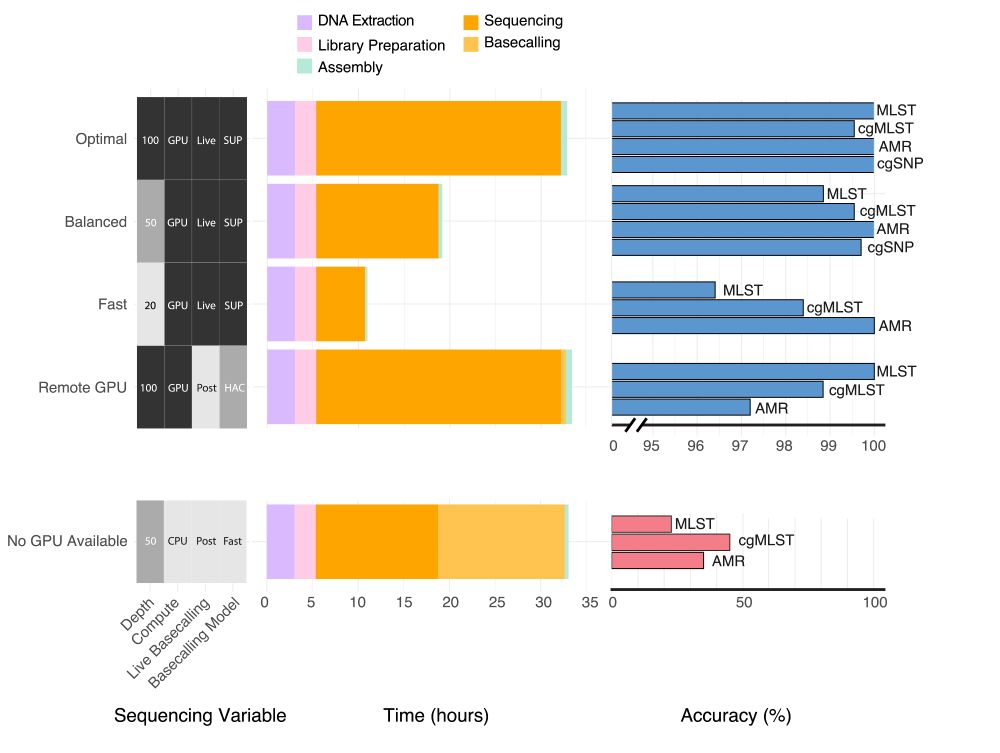
Ryan Wick
@rrwick.bsky.social
· Jun 10
Ryan Wick
@rrwick.bsky.social
· May 30
Ryan Wick
@rrwick.bsky.social
· May 30
Ryan Wick
@rrwick.bsky.social
· May 30