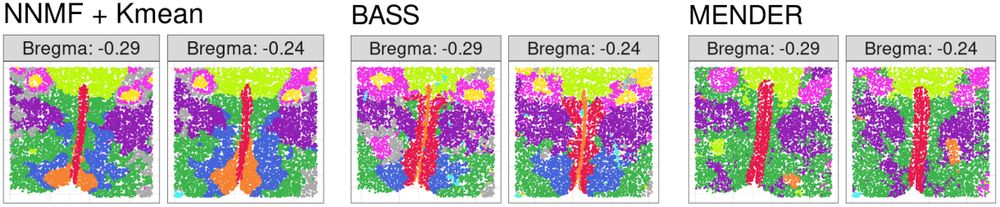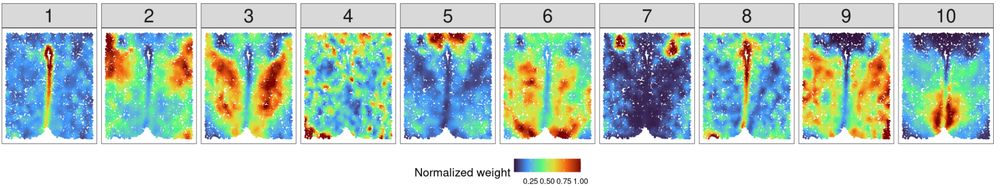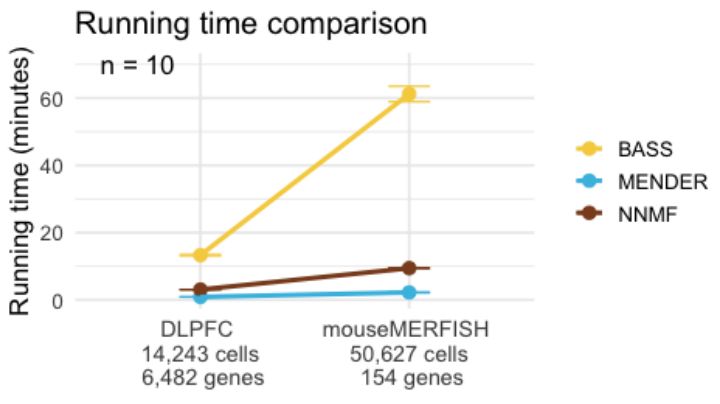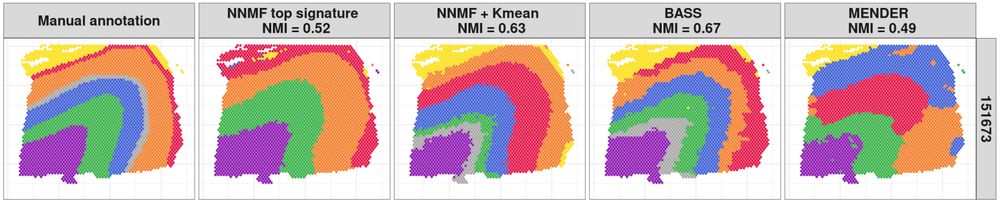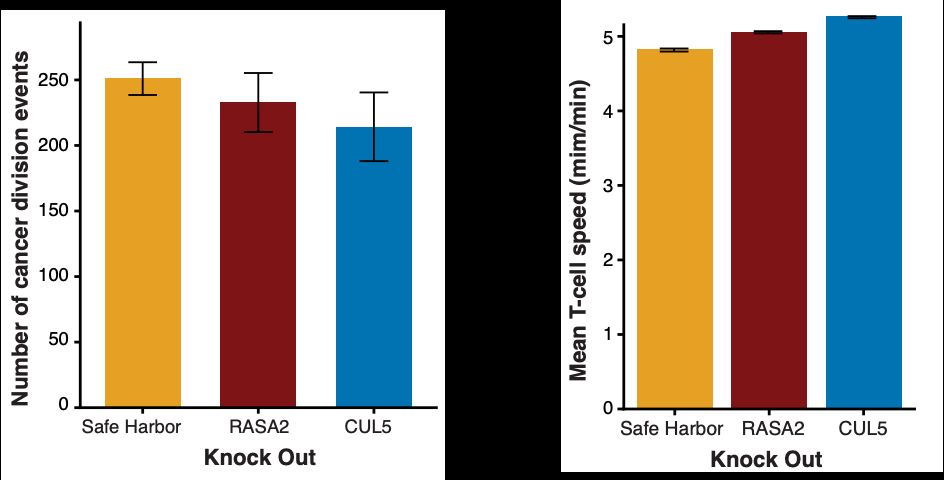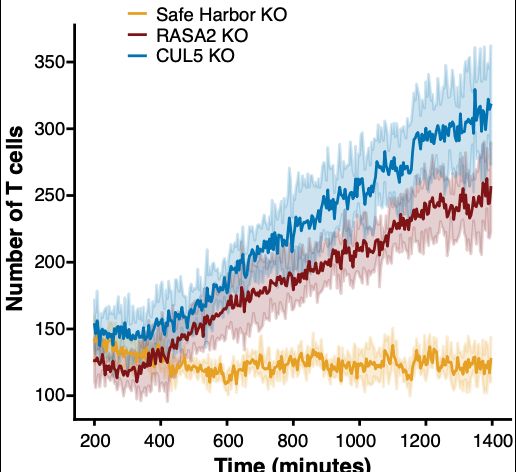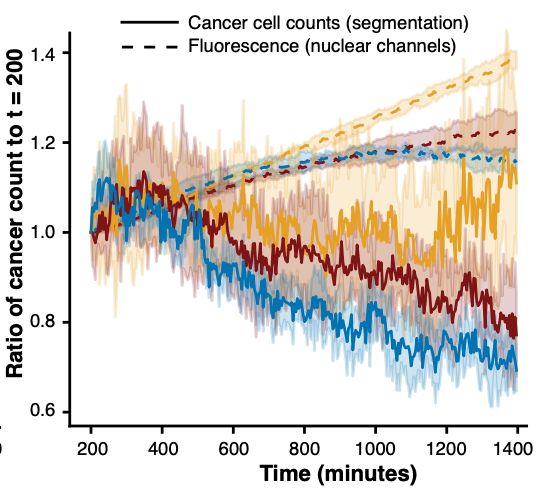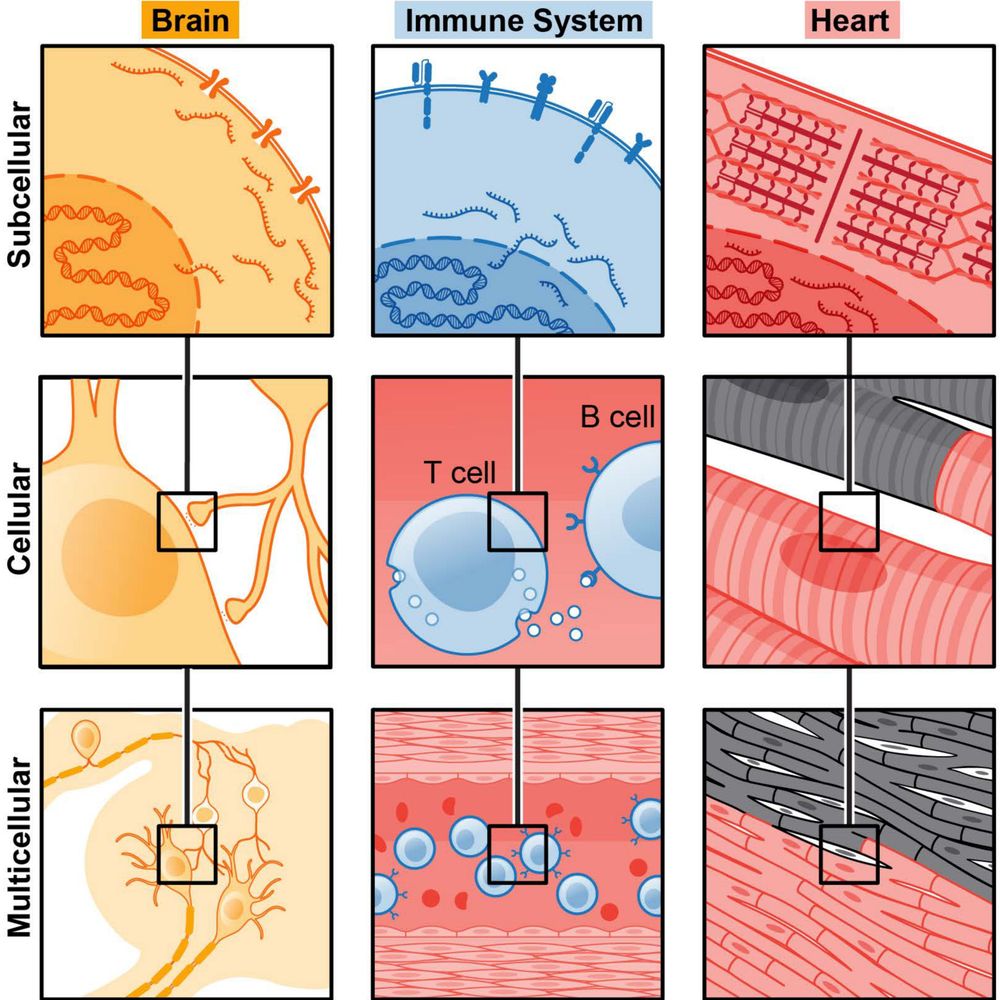
Engelhardt Research Group
@thebeehive.bsky.social
340 followers
590 following
33 posts
Engelhardt Research Group at Stanford University and Gladstone Institutes. Statistical genomics, live-cell imaging, wearable data, cancer immunology, reproductive health.
Posts
Media
Videos
Starter Packs