
Hannah Wayment-Steele
@hkws.bsky.social
1.1K followers
98 following
49 posts
Avid rower who sometimes thinks about biomolecular dynamics. Asst Prof @uwbiochem.bsky.social
https://waymentsteelelab.org/
Posts
Media
Videos
Starter Packs



Hannah Wayment-Steele
@hkws.bsky.social
· May 23

Scientist and Lab Operations Coordinator - Madison, Wisconsin, United States
Job Summary:
The Wayment-Steele Lab seeks a Research Scientist to lead experimental operations within our highly interdisciplinary team, integrating both computational "dry" and experimental "wet" l...
jobs.wisc.edu
Reposted by Hannah Wayment-Steele


Reposted by Hannah Wayment-Steele

Reposted by Hannah Wayment-Steele






Hannah Wayment-Steele
@hkws.bsky.social
· Apr 28



Reposted by Hannah Wayment-Steele

Reposted by Hannah Wayment-Steele

Rita Strack
@ritastrack.bsky.social
· Apr 10
Hannah Wayment-Steele
@hkws.bsky.social
· Mar 21
Hannah Wayment-Steele
@hkws.bsky.social
· Mar 21
Reposted by Hannah Wayment-Steele





Reposted by Hannah Wayment-Steele
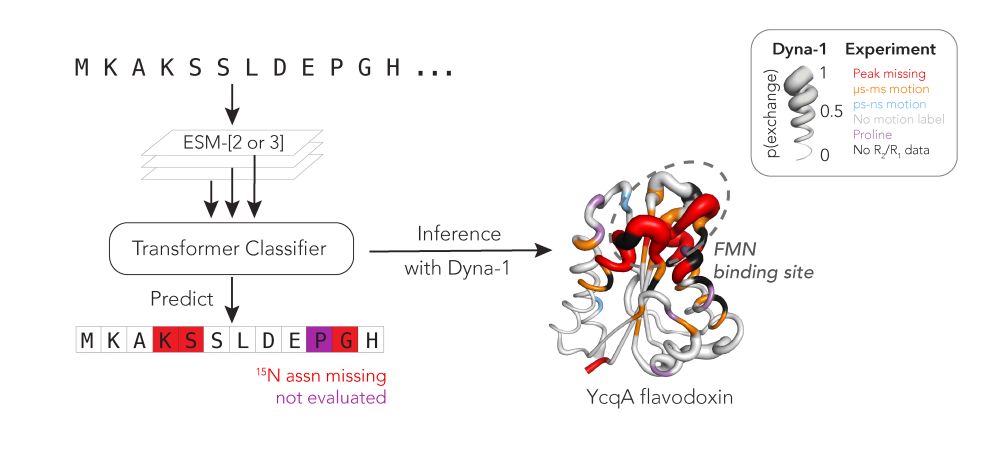
Hannah Wayment-Steele
@hkws.bsky.social
· Mar 20
Hannah Wayment-Steele
@hkws.bsky.social
· Mar 20

Hannah Wayment-Steele
@hkws.bsky.social
· Mar 20
