James Hemker
@jahemker.bsky.social
77 followers
88 following
17 posts
Stanford Dev Bio PhD
@petrovadmitri.bsky.social's lab
Carcharodontosaurus is my favorite dinosaur.
Posts
Media
Videos
Starter Packs



James Hemker
@jahemker.bsky.social
· Jun 21

Reposted by James Hemker

Ben Moran
@ben-moran.bsky.social
· May 1

Increased rates of hybridization in swordtail fish are associated with water pollution
Biodiversity loss can occur when disturbance compromises the reproductive barriers between species, causing them to collapse into a single population through hybridization. Recent research has documen...
www.biorxiv.org
James Hemker
@jahemker.bsky.social
· Apr 25
James Hemker
@jahemker.bsky.social
· Apr 25


James Hemker
@jahemker.bsky.social
· Apr 25
James Hemker
@jahemker.bsky.social
· Apr 25
James Hemker
@jahemker.bsky.social
· Apr 25


James Hemker
@jahemker.bsky.social
· Apr 25
James Hemker
@jahemker.bsky.social
· Apr 25
James Hemker
@jahemker.bsky.social
· Apr 25
James Hemker
@jahemker.bsky.social
· Apr 25
James Hemker
@jahemker.bsky.social
· Apr 25

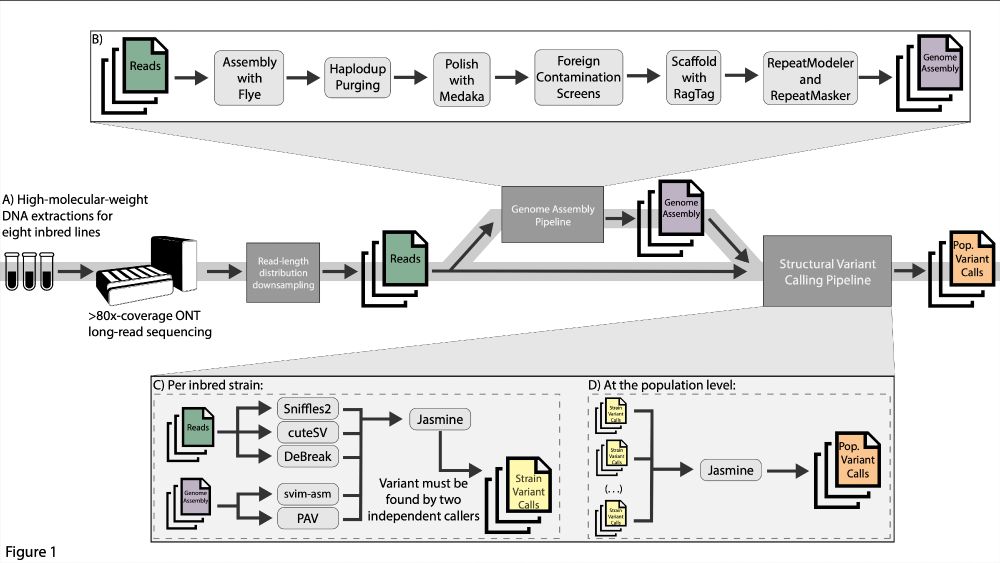
Manual validation finds only ultra-long long-read sequencing enables faithful, population-level structural variant calling in Drosophila melanogaster euchromatin
The increasing accessibility of long-read sequencing and the rapid development of automated variant callers are promoting the generation of population-level structural variation data. However, the eff...
doi.org
Reposted by James Hemker
