James Lingford
@jameslingford.bsky.social
160 followers
230 following
60 posts
PhD student in structural biology with @greening.bsky.social and @knottrna.bsky.social at Monash Uni. (he/him)
Interested in hydrogenases, evolution, protein design.
💻 https://www.jameslingford.com/
Posts
Media
Videos
Starter Packs





James Lingford
@jameslingford.bsky.social
· Aug 27

Large protein databases reveal structural complementarity and functional locality - Nature Communications
Researchers mapped the protein structure landscape, revealing structural complementarity across databases and functional clustering in specific regions. Their web tool helps explore this space, unlock...
www.nature.com
Reposted by James Lingford


James Lingford
@jameslingford.bsky.social
· Aug 21

GitHub - lehner-lab/combinatorialcores: Source code for analyses and figure reproduction in "Genetics, energetics, and allostery in proteins with randomized cores and surfaces", Escobedo et. al Scienc...
Source code for analyses and figure reproduction in "Genetics, energetics, and allostery in proteins with randomized cores and surfaces", Escobedo et. al Science 2025 - lehner-lab/combina...
github.com
James Lingford
@jameslingford.bsky.social
· Aug 13
James Lingford
@jameslingford.bsky.social
· Aug 13

Will Ratcliff
@wcratcliff.bsky.social
· Aug 12

Reposted by James Lingford


Reposted by James Lingford

Reposted by James Lingford



Reposted by James Lingford


Reposted by James Lingford

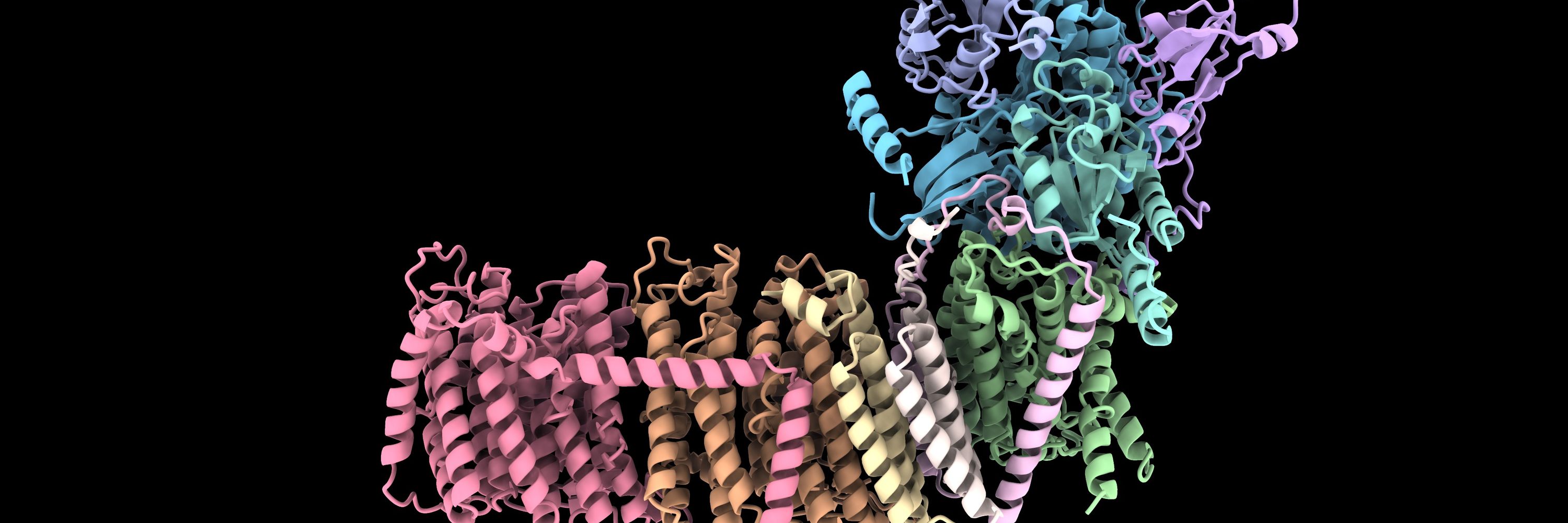
Rhys Grinter
@rhyswg.bsky.social
· Jul 9
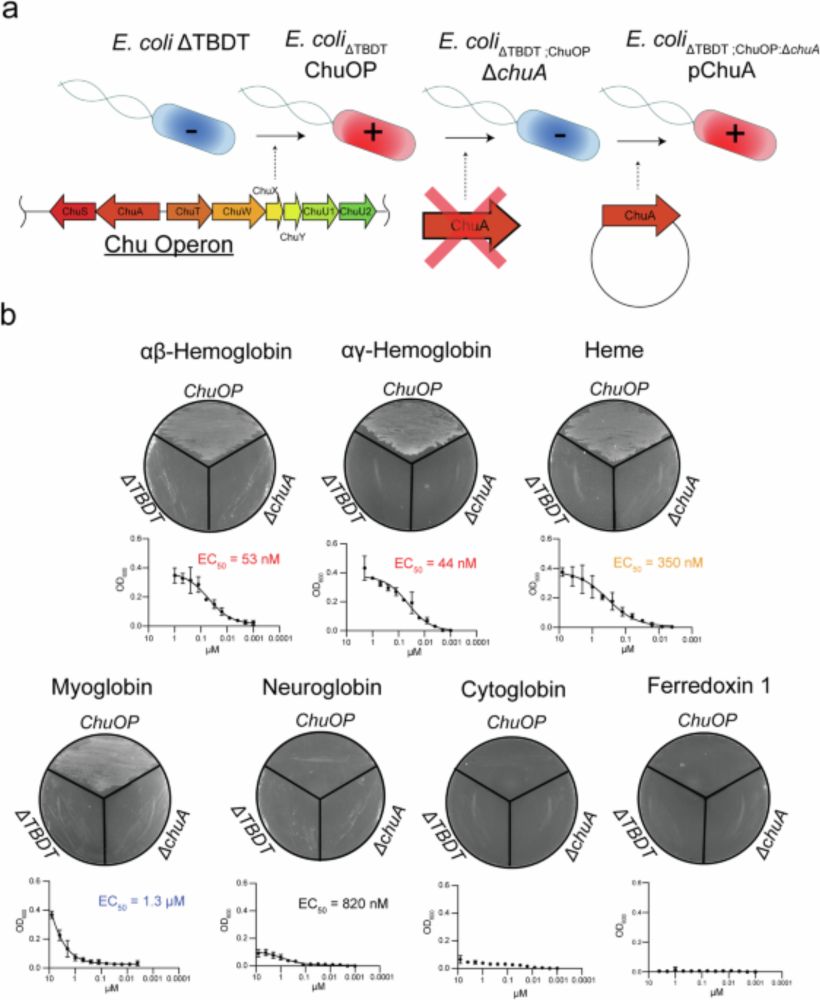
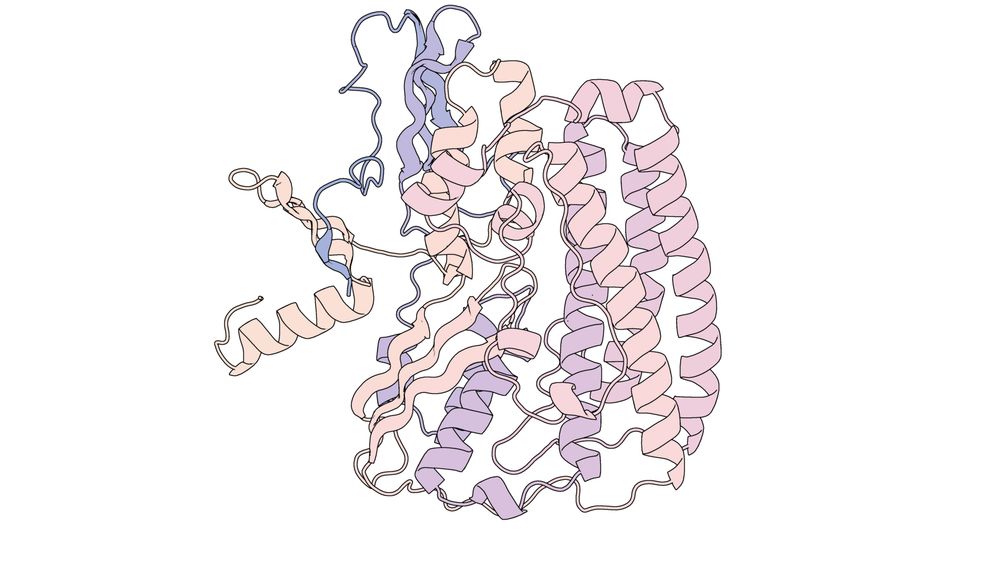
Inhibiting heme piracy by pathogenic Escherichia coli using de novo-designed proteins - Nature Communications
Many pathogens encode transporters that extract heme directly from host proteins. In this study, the authors demonstrate the utility of de novo-designed proteins in understanding the mechanism behind ...
www.nature.com
Reposted by James Lingford

Reposted by James Lingford