
Jeff Spence
@jeffspence.github.io
980 followers
730 following
86 posts
assistant professor at ucsf interested in genetics, statistics, etc…
jeffspence.github.io
Posts
Media
Videos
Starter Packs
Pinned

Jeff Spence
@jeffspence.github.io
· Dec 17

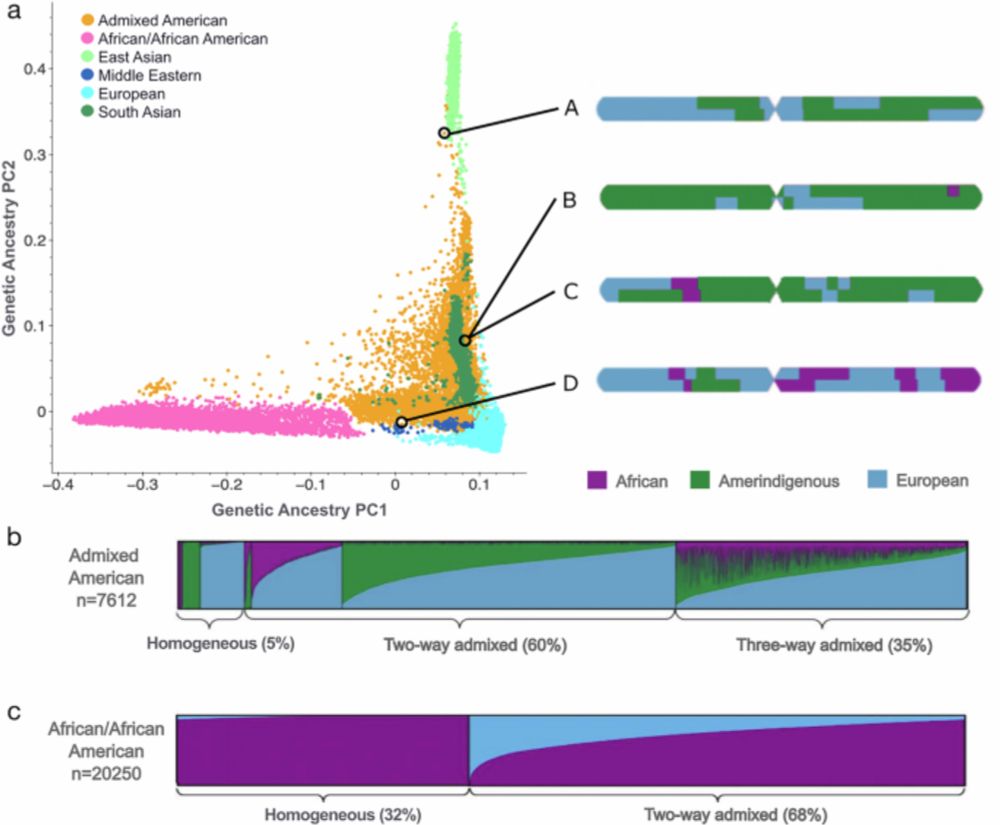
Specificity, length, and luck: How genes are prioritized by rare and common variant association studies
Standard genome-wide association studies (GWAS) and rare variant burden tests are essential tools for identifying trait-relevant genes. Although these methods are conceptually similar, we show by anal...
www.biorxiv.org

Reposted by Jeff Spence




Reposted by Jeff Spence



Reposted by Jeff Spence


Reposted by Jeff Spence


Reposted by Jeff Spence

Reposted by Jeff Spence

Reposted by Jeff Spence
Reposted by Jeff Spence

Reposted by Jeff Spence

Reposted by Jeff Spence






Reposted by Jeff Spence

Reposted by Jeff Spence


Reposted by Jeff Spence


Jeff Spence
@jeffspence.github.io
· Sep 7
Jeff Spence
@jeffspence.github.io
· Sep 5
Jeff Spence
@jeffspence.github.io
· Sep 5