Kieran Didi
@kdidi.bsky.social
1.1K followers
200 following
14 posts
🧪 Research Scientist @nvidia and PhD student @Oxford staring at proteins all-day
🧑💻 Website/Blog: https://kdidi.netlify.app/
🤖 GitHub: https://github.com/kierandidi
📚 Prev. Cambridge/Heidelberg
Posts
Media
Videos
Starter Packs




Kieran Didi
@kdidi.bsky.social
· Aug 15

Nate Corley
@ncorley.bsky.social
· Aug 15

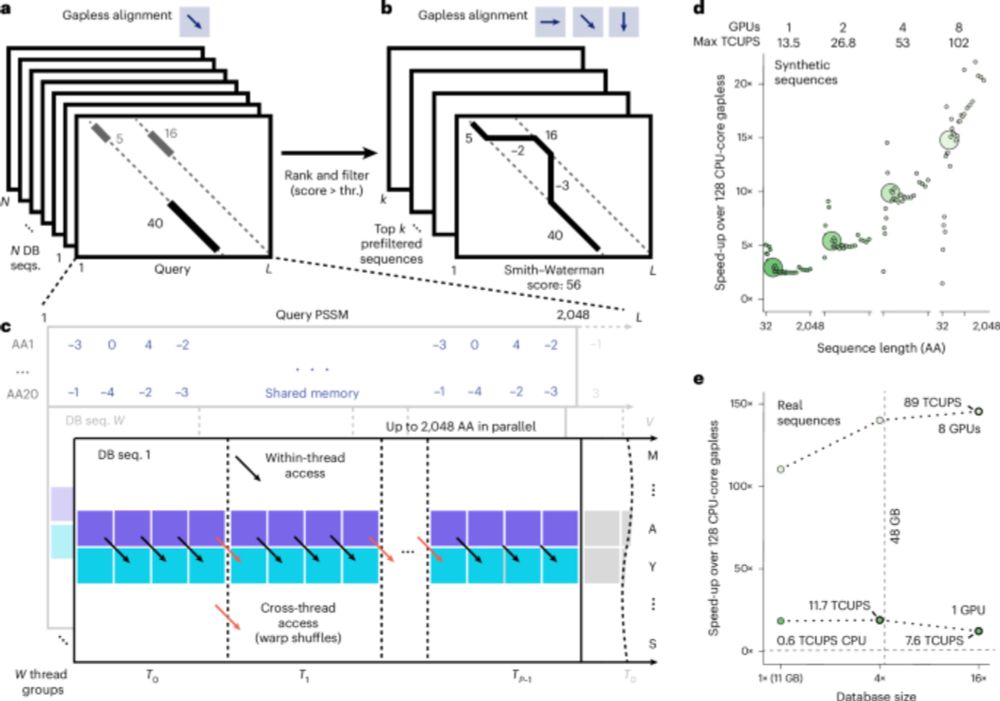
Accelerating Biomolecular Modeling with AtomWorks and RF3
Deep learning methods trained on protein structure databases have revolutionized biomolecular structure prediction, but developing and training new models remains a considerable challenge. To facilita...
www.biorxiv.org
Kieran Didi
@kdidi.bsky.social
· Aug 15




Reposted by Kieran Didi


Reposted by Kieran Didi
Nate Corley
@ncorley.bsky.social
· Aug 15
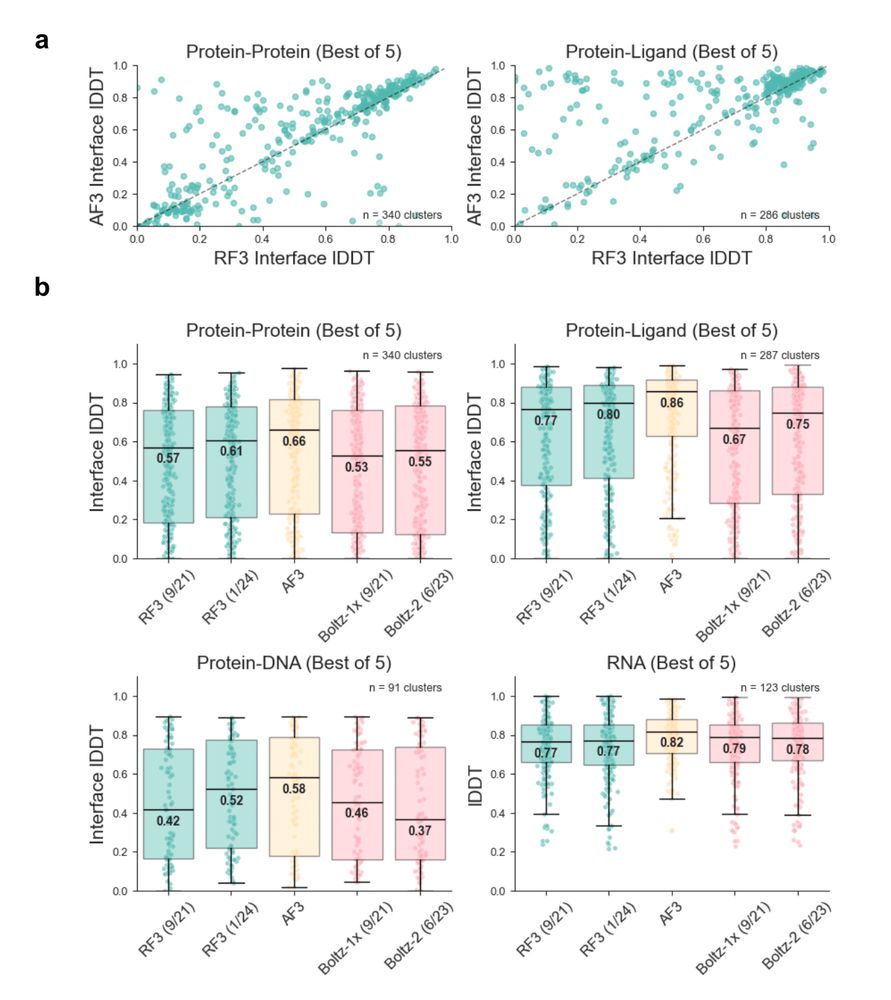
Accelerating Biomolecular Modeling with AtomWorks and RF3
Deep learning methods trained on protein structure databases have revolutionized biomolecular structure prediction, but developing and training new models remains a considerable challenge. To facilita...
www.biorxiv.org
Kieran Didi
@kdidi.bsky.social
· Jul 19

Reposted by Kieran Didi

Anshul Kundaje
@anshulkundaje.bsky.social
· Feb 20

Reposted by Kieran Didi

Reposted by Kieran Didi

Kieran Didi
@kdidi.bsky.social
· Dec 23

Reposted by Kieran Didi




Reposted by Kieran Didi


estella newcombe
@estellaan.bsky.social
· Nov 27
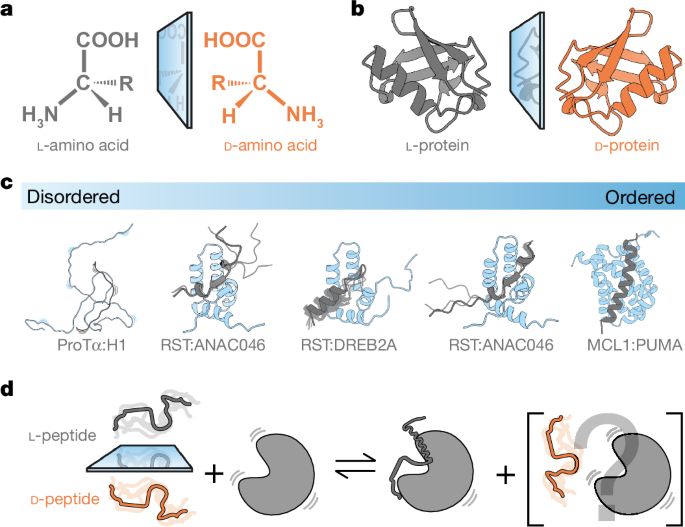
Stereochemistry in the disorder–order continuum of protein interactions - Nature
Studies on protein–protein interactions using proteins containing d- or l-amino acids show that stereoselectivity of binding varies with the degree of disorder within the complex.
www.nature.com
Kieran Didi
@kdidi.bsky.social
· Nov 16