Konrad
@konradjk.bsky.social
1.2K followers
44 following
25 posts
Genomicist, computational biologist. Assistant professor @ MGH, HMS. Associate member @ Broad Institute
https://klab.is
Posts
Media
Videos
Starter Packs

Konrad
@konradjk.bsky.social
· 12h
Konrad
@konradjk.bsky.social
· 20d
Konrad
@konradjk.bsky.social
· 20d
Konrad
@konradjk.bsky.social
· 20d

Pan-UK Biobank genome-wide association analyses enhance discovery and resolution of ancestry-enriched effects - Nature Genetics
Genome-wide analyses for 7,266 traits leveraging data from several genetic ancestry groups in UK Biobank identify new associations and enhance resources for interpreting risk variants across diverse p...
www.nature.com
Konrad
@konradjk.bsky.social
· Nov 8
Konrad
@konradjk.bsky.social
· Nov 7
Konrad
@konradjk.bsky.social
· Sep 21
Konrad
@konradjk.bsky.social
· Sep 20
Konrad
@konradjk.bsky.social
· Sep 20

Konrad
@konradjk.bsky.social
· Sep 20
Reposted by Konrad

Kaitlin Samocha
@ksamocha.bsky.social
· Apr 19

Konrad
@konradjk.bsky.social
· Dec 7
Konrad
@konradjk.bsky.social
· Dec 7
Konrad
@konradjk.bsky.social
· Dec 6
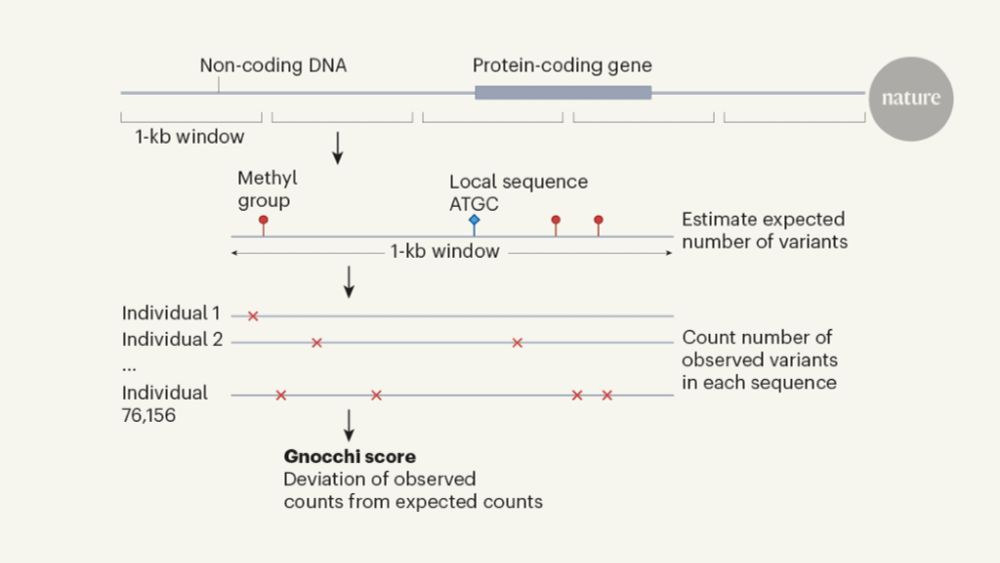
An expanded genomic database for identifying disease-related variants
An expanded version of a human-genome database called gnomAD, containing 76,156 whole-genome sequences, has enabled investigation of how variants in non-protein-coding regions of the genome affect hea...
www.nature.com
Konrad
@konradjk.bsky.social
· Dec 6
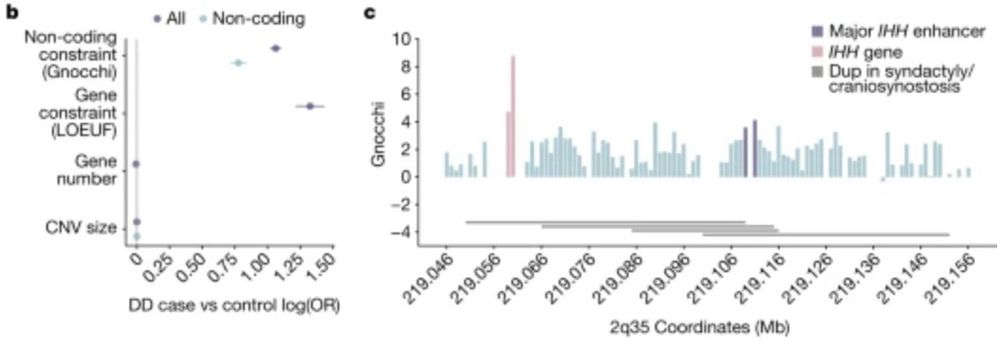
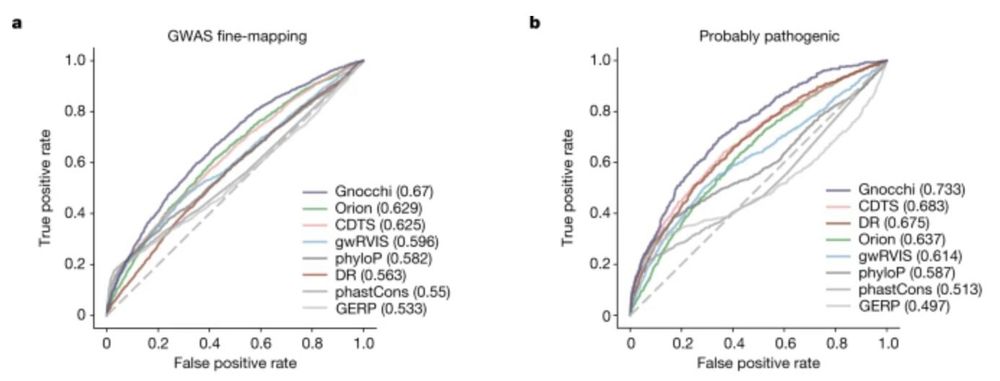
Konrad
@konradjk.bsky.social
· Dec 6
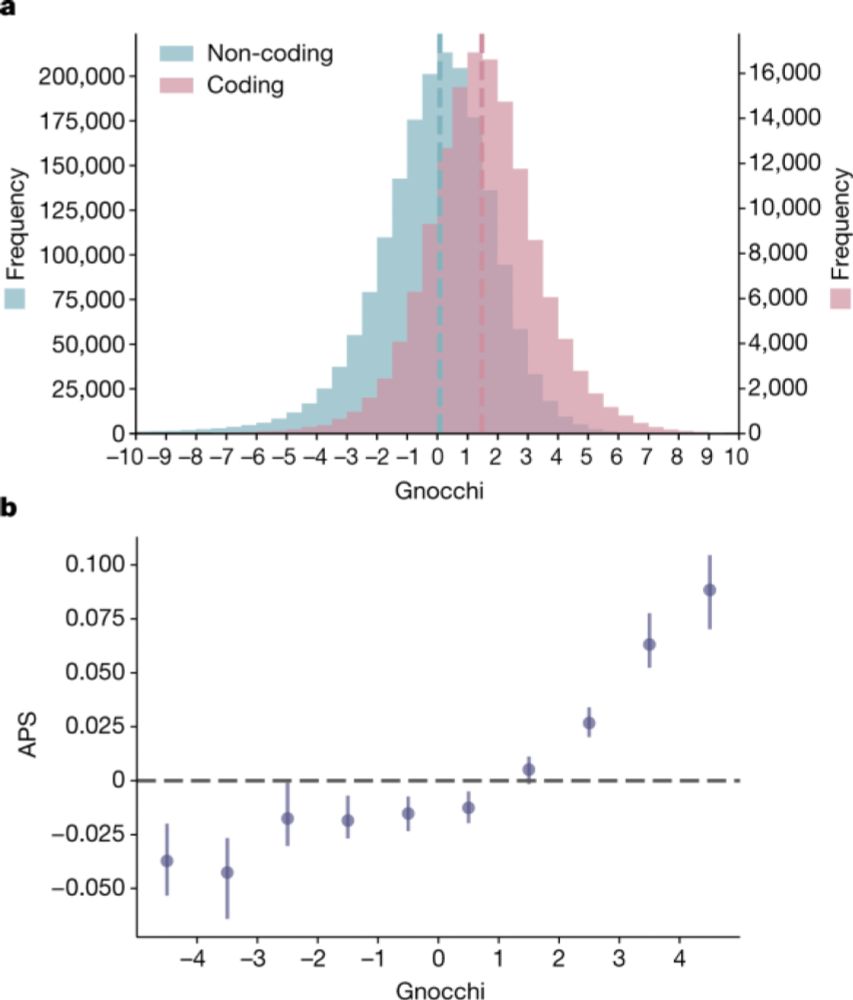
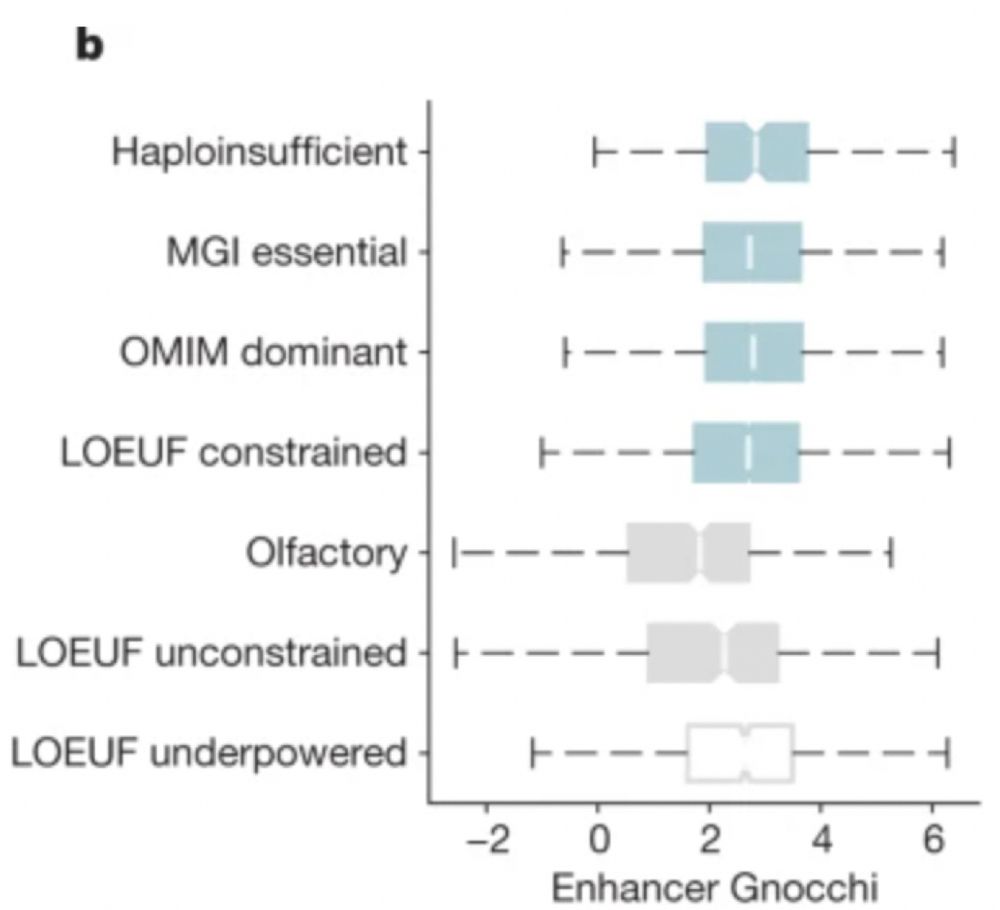
A genomic mutational constraint map using variation in 76,156 human genomes - Nature
A genomic constraint map for the human genome constructed using data from 76,156 human genomes from the Genome Aggregation Database shows that non-coding constrained regions are enriched for regulator...
idp.nature.com