Kaitlin Samocha
@ksamocha.bsky.social
1.5K followers
230 following
58 posts
Assistant Investigator @ MGH / Broad / HMS. Focus on human genomics and modeling rare variation. She/her
Posts
Media
Videos
Starter Packs

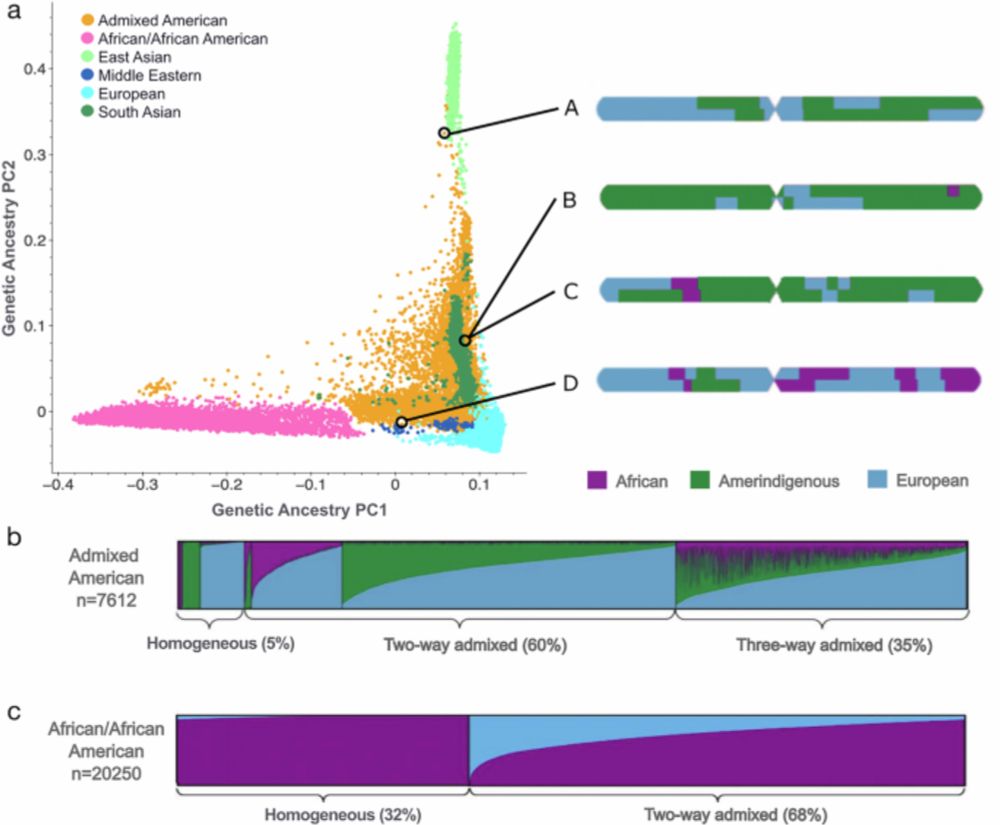


Reposted by Kaitlin Samocha


Reposted by Kaitlin Samocha

Reposted by Kaitlin Samocha




Reposted by Kaitlin Samocha

Alex Hoischen
@ahoischen.bsky.social
· May 24
Reposted by Kaitlin Samocha

Zornitza Stark
@zornitza.bsky.social
· May 23

Scalable automated reanalysis of genomic data in research and clinical rare disease cohorts
Reanalysis of genomic data in rare disease is highly effective in increasing diagnostic yields but remains limited by manual approaches. Automation and optimization for high specificity will be necess...
www.medrxiv.org
Reposted by Kaitlin Samocha

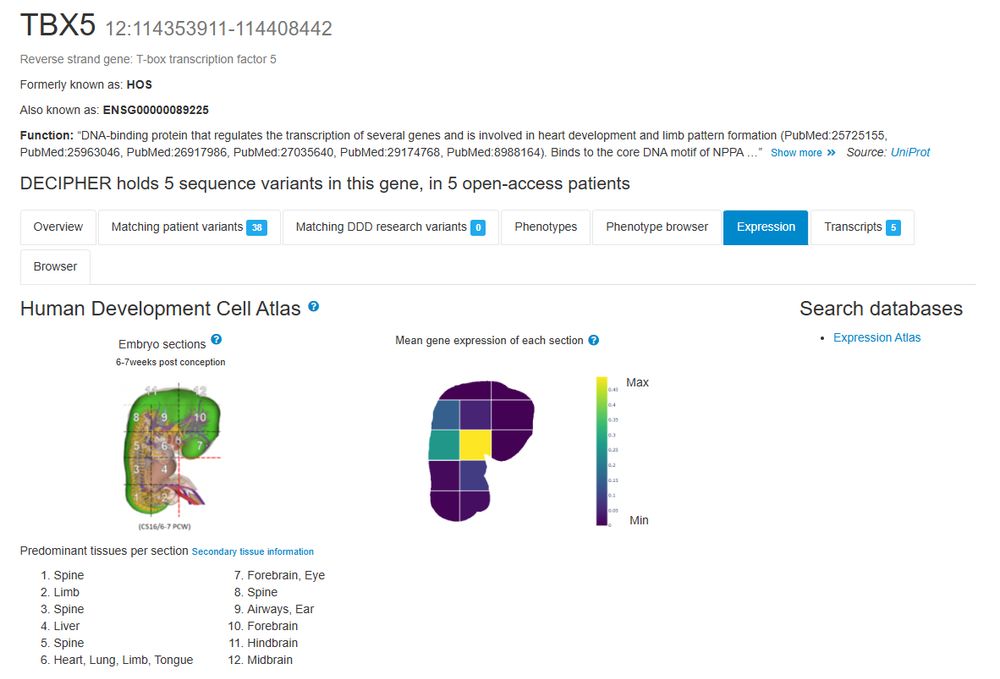

Kaitlin Samocha
@ksamocha.bsky.social
· Feb 21
Reposted by Kaitlin Samocha


Kaitlin Samocha
@ksamocha.bsky.social
· Apr 19


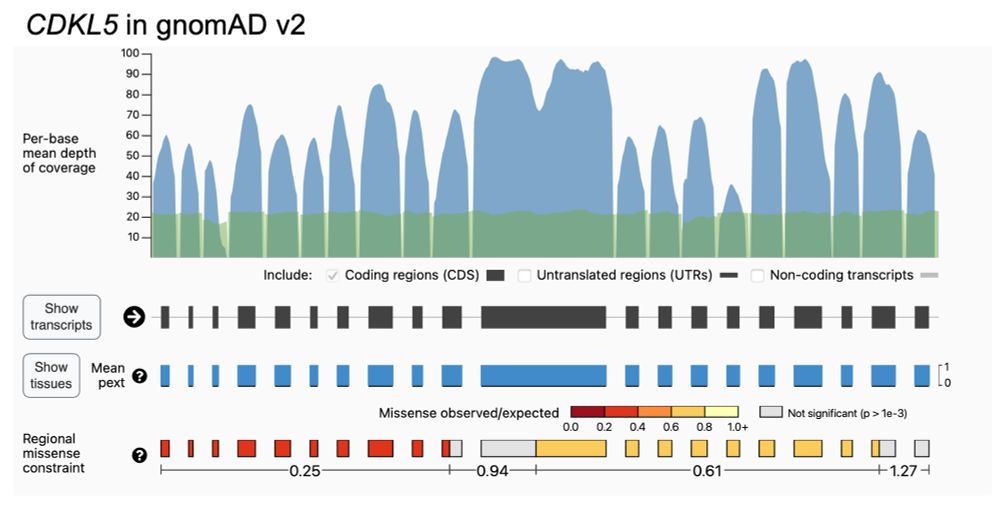
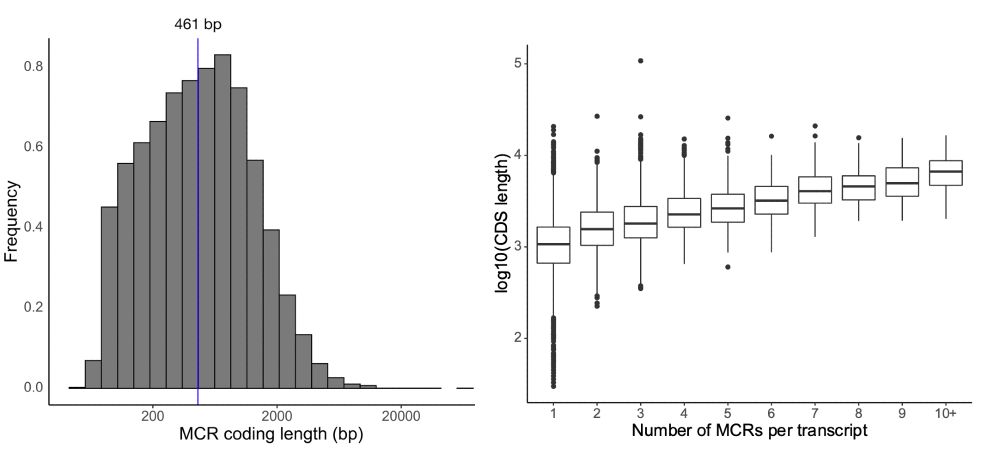
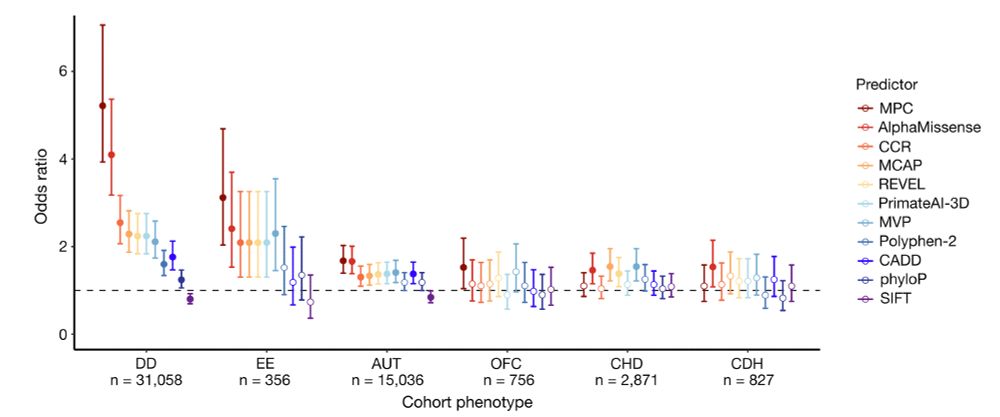

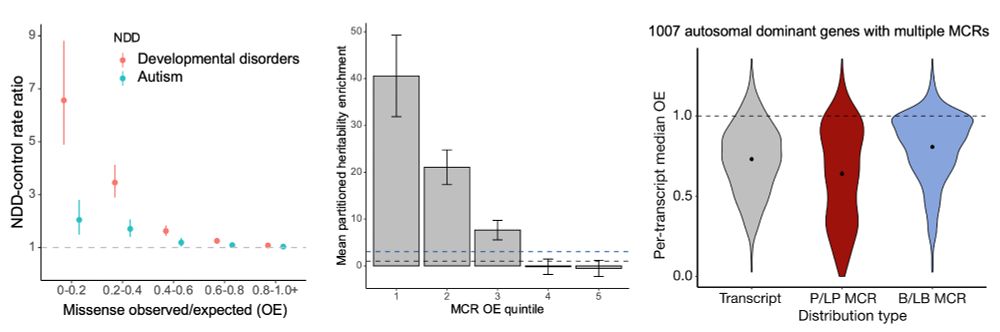
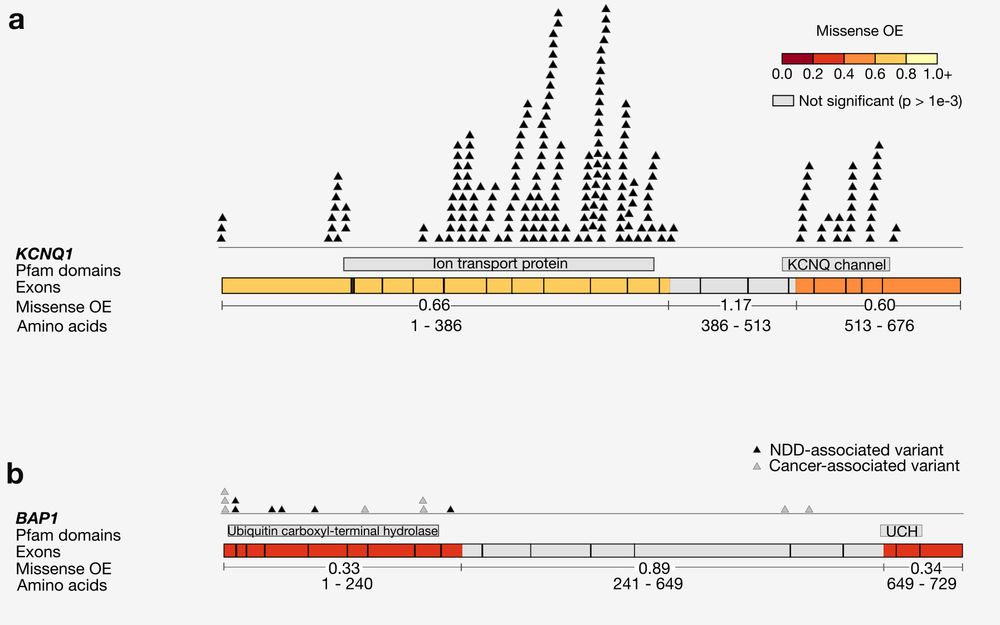
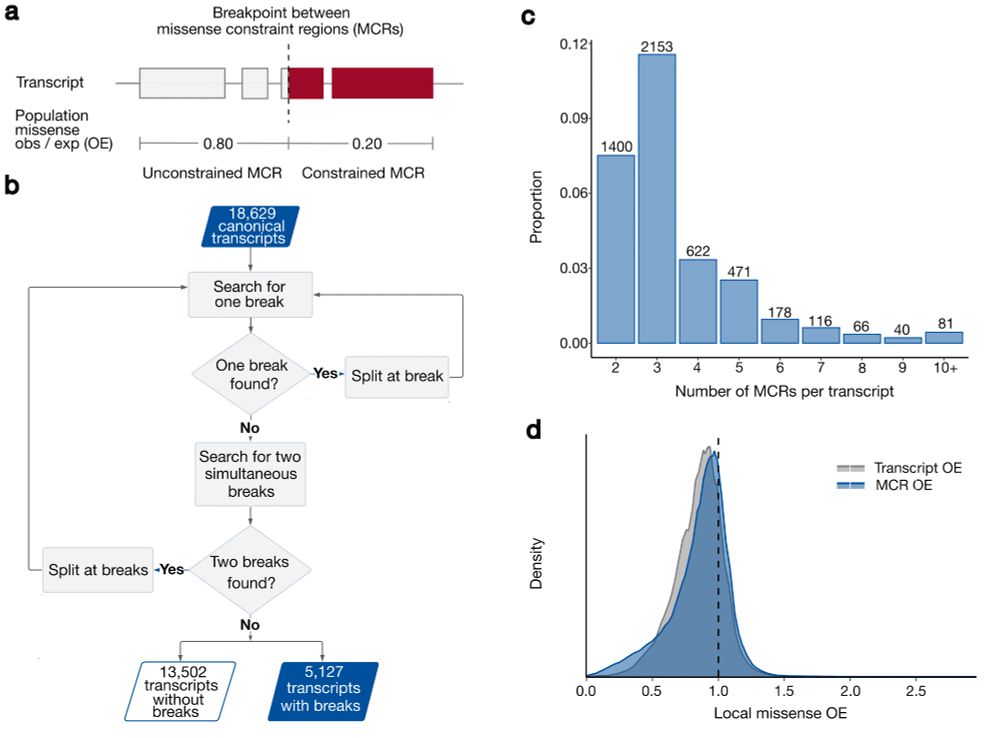
Kaitlin Samocha
@ksamocha.bsky.social
· Apr 19
Kaitlin Samocha
@ksamocha.bsky.social
· Mar 8
Kaitlin Samocha
@ksamocha.bsky.social
· Dec 8
Kaitlin Samocha
@ksamocha.bsky.social
· Dec 8