Laurits Skov
@lauritsskov.bsky.social
410 followers
310 following
29 posts
I study Neanderthals, Denisovans and the effect of their DNA surviving in present day humans. Assistant professor at section for molecular ecology and evolution at Globe Institute Copenhagen, Denmark.
Posts
Media
Videos
Starter Packs




Reposted by Laurits Skov


Laurits Skov
@lauritsskov.bsky.social
· May 5
Laurits Skov
@lauritsskov.bsky.social
· May 5

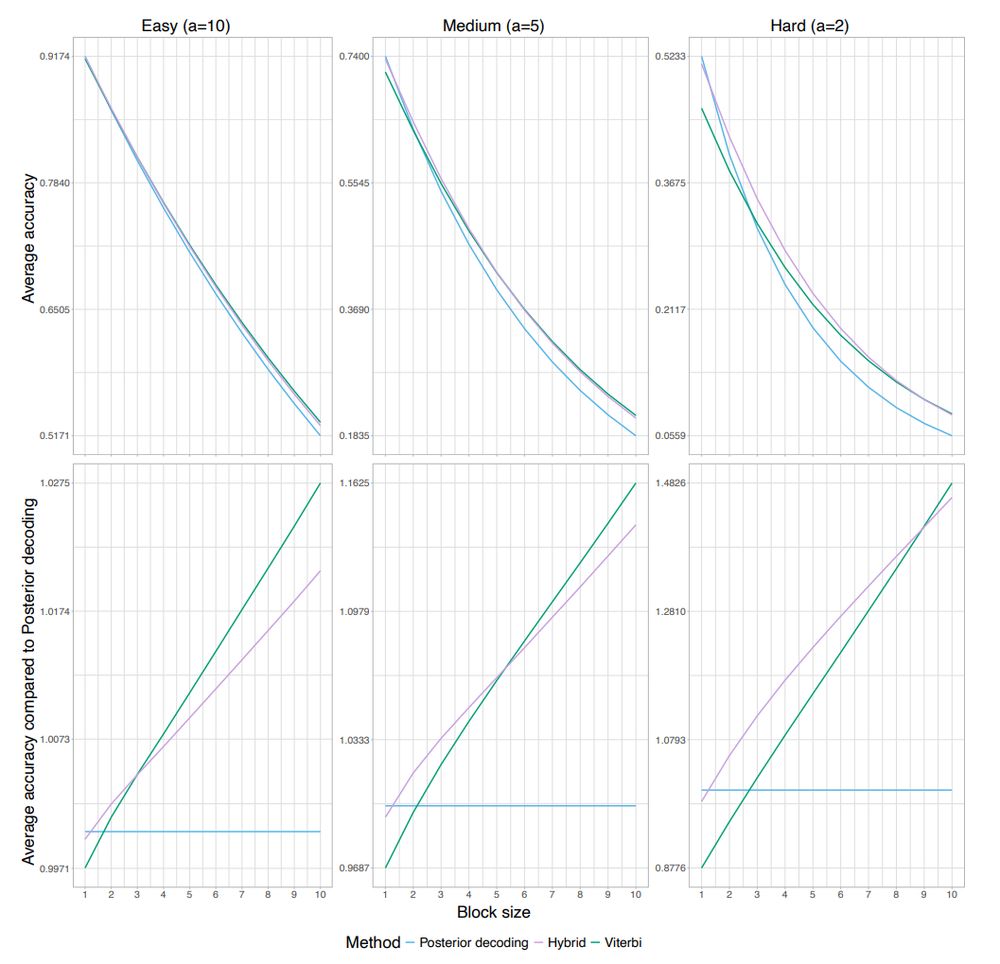

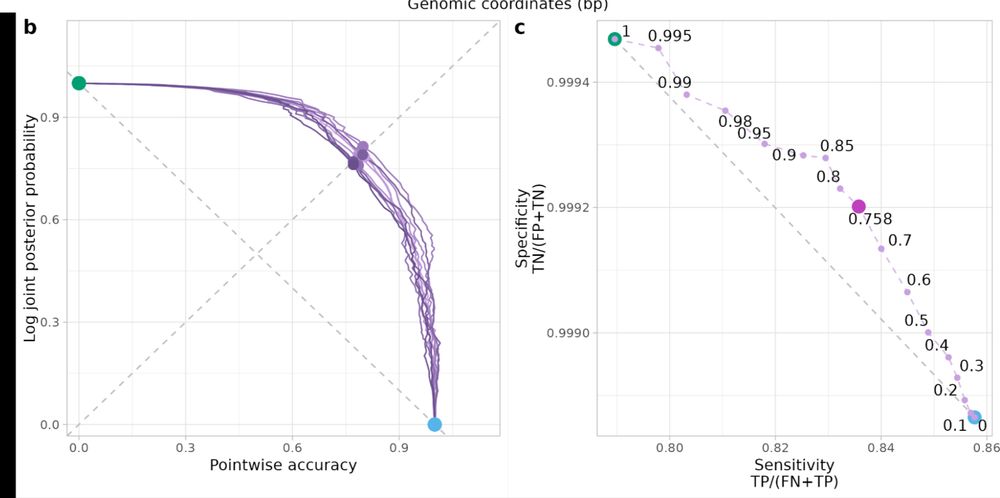
Laurits Skov
@lauritsskov.bsky.social
· May 5

Laurits Skov
@lauritsskov.bsky.social
· May 5
Laurits Skov
@lauritsskov.bsky.social
· May 5
Laurits Skov
@lauritsskov.bsky.social
· May 5
Laurits Skov
@lauritsskov.bsky.social
· May 2
Laurits Skov
@lauritsskov.bsky.social
· May 1
Laurits Skov
@lauritsskov.bsky.social
· May 1
Laurits Skov
@lauritsskov.bsky.social
· May 1
Laurits Skov
@lauritsskov.bsky.social
· May 1




Laurits Skov
@lauritsskov.bsky.social
· May 1