Mathieu Linares
@mathieulinares.bsky.social
35 followers
56 following
14 posts
Application Expert at PDC @KTH Working on the interoperability of #compchem tools. Active developer of @veloxchem.bsky.social and @viamd.bsky.social
Posts
Media
Videos
Starter Packs
Pinned

Reposted by Mathieu Linares



Reposted by Mathieu Linares


Reposted by Mathieu Linares

VeloxChem
@veloxchem.bsky.social
· Aug 7

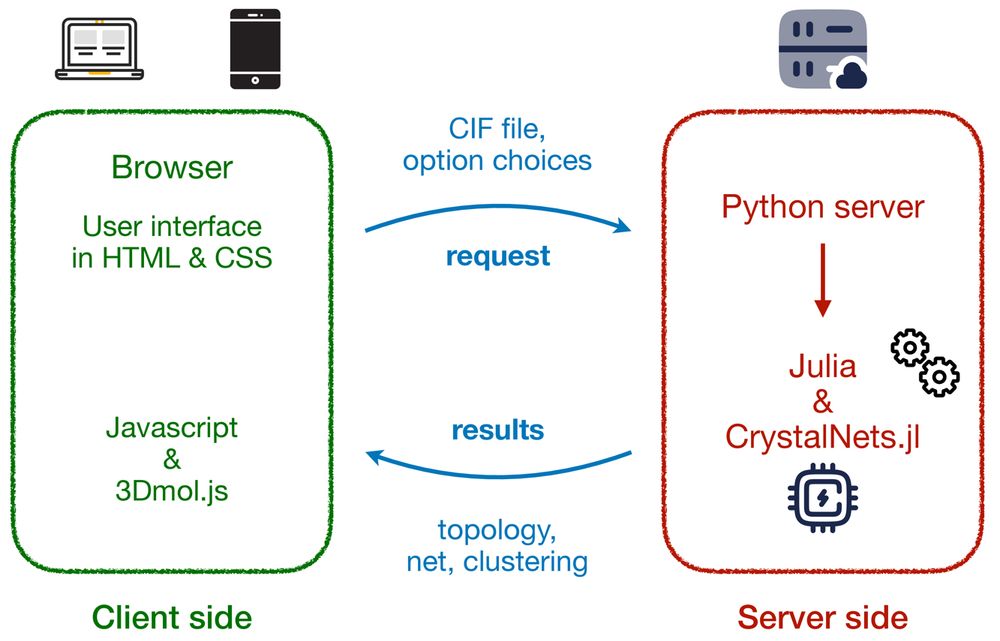
VeloxChem Quantum–Classical Interoperability for Modeling of Complex Molecular Systems
Being a program written primarily in Python that strictly adheres to modern object-oriented software engineering and parallel programming practices, VeloxChem is shown to be suitable for the development of (semi)automatized workflows that extend its scope from first-principles quantum chemical purism to hybrid quantum–classical interoperability and some degree of semiempiricism. Methods are presented for building complex systems such as metal–organic frameworks, constructing molecular mechanics and interpolation mechanics force fields, conformer searches, system solvation, determining free energies of solvation, and determining free energy profiles of reaction pathways using the empirical valence bond method. The implementations are made intuitive with opportunities for interactive plotting and 3D molecular structure illustrations through the use of Jupyter notebooks.
doi.org
Reposted by Mathieu Linares

VIAMD
@viamd.bsky.social
· Aug 7
VeloxChem
@veloxchem.bsky.social
· Aug 7
VeloxChem Quantum–Classical Interoperability for Modeling of Complex Molecular Systems
Being a program written primarily in Python that strictly adheres to modern object-oriented software engineering and parallel programming practices, VeloxChem is shown to be suitable for the development of (semi)automatized workflows that extend its scope from first-principles quantum chemical purism to hybrid quantum–classical interoperability and some degree of semiempiricism. Methods are presented for building complex systems such as metal–organic frameworks, constructing molecular mechanics and interpolation mechanics force fields, conformer searches, system solvation, determining free energies of solvation, and determining free energy profiles of reaction pathways using the empirical valence bond method. The implementations are made intuitive with opportunities for interactive plotting and 3D molecular structure illustrations through the use of Jupyter notebooks.
doi.org
Reposted by Mathieu Linares

Reposted by Mathieu Linares

Reposted by Mathieu Linares

Reposted by Mathieu Linares





Reposted by Mathieu Linares

Reposted by Mathieu Linares
Reposted by Mathieu Linares
BioExcel CoE
@bioexcelcoe.bsky.social
· Jun 4
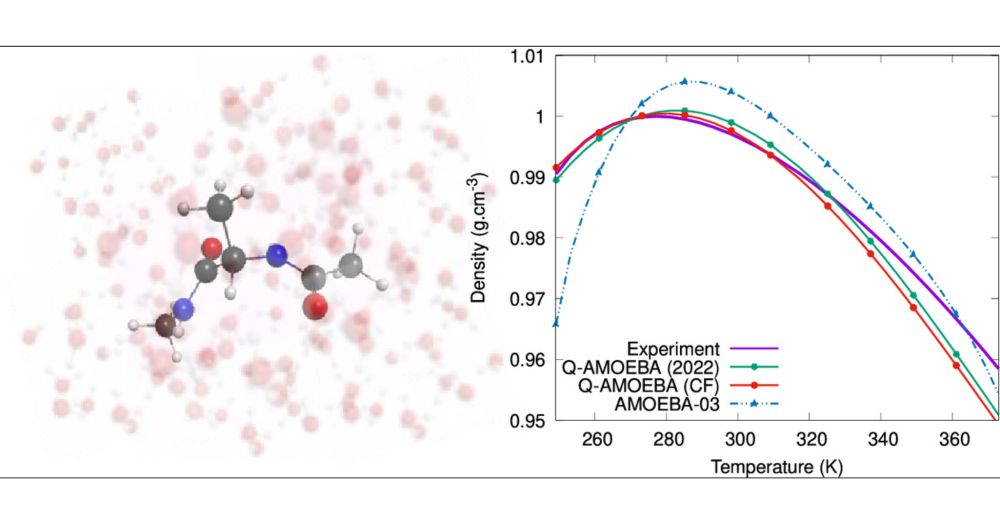
Reposted by Mathieu Linares
