
Melé Lab
@melelab.bsky.social
150 followers
250 following
24 posts
We are the Melé Lab, the Transcriptomics and Functional Genomics Lab (TFGL) at the Barcelona Supercomputing Center (BSC).
Account by lab members
Posts
Media
Videos
Starter Packs




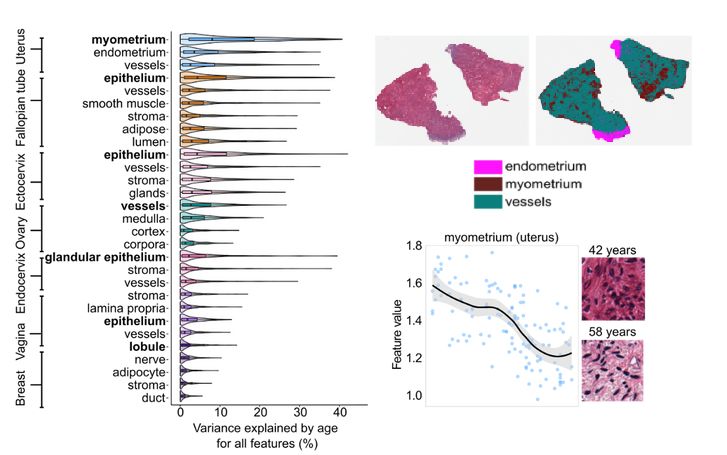


Melé Lab
@melelab.bsky.social
· May 23
Reposted by Melé Lab
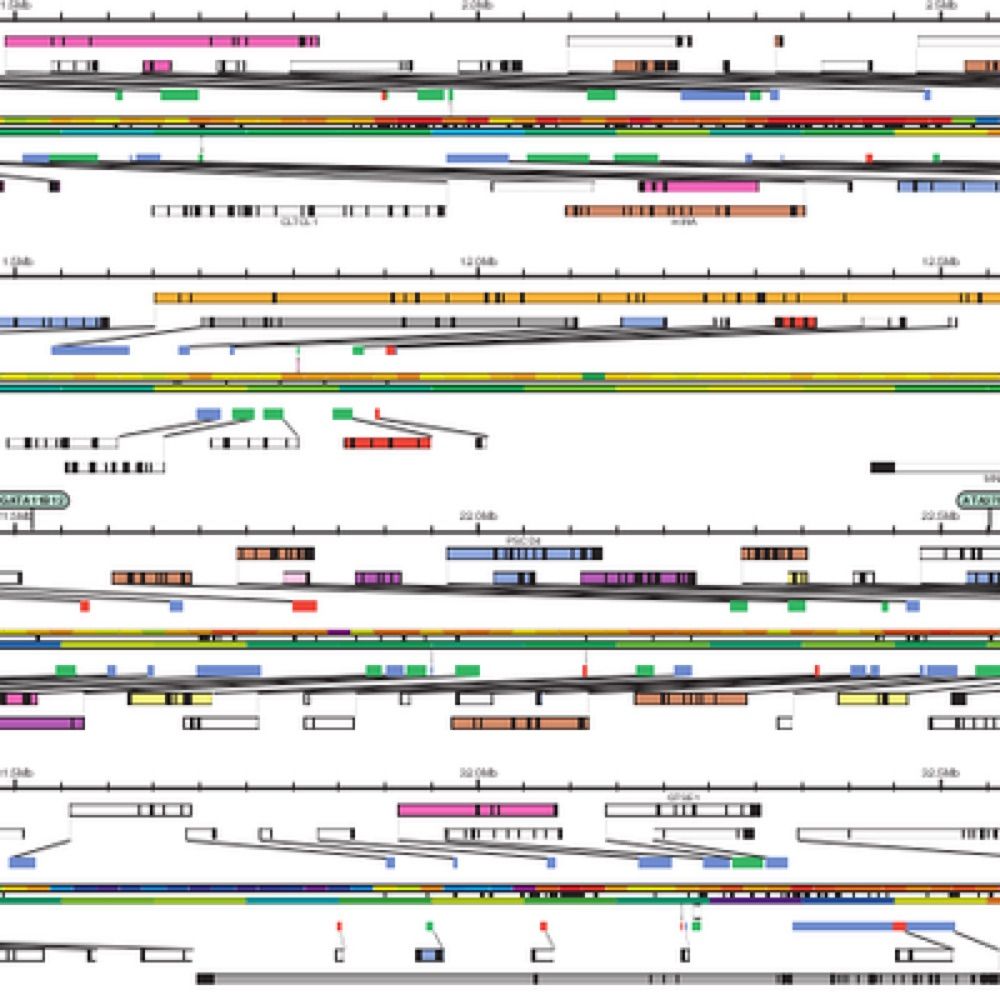
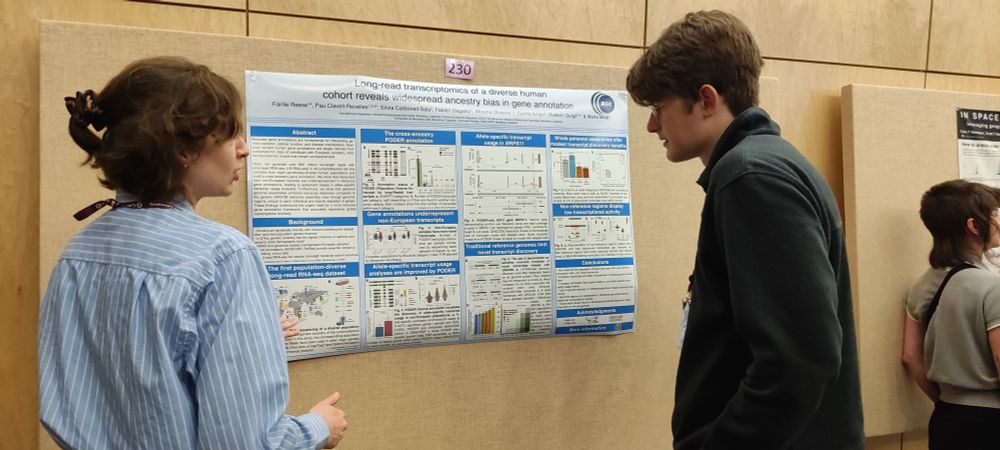



Melé Lab
@melelab.bsky.social
· Mar 20
Melé Lab
@melelab.bsky.social
· Mar 20

Long-read transcriptomics of a diverse human cohort reveals widespread ancestry bias in gene annotation
Accurate gene annotations are fundamental for interpreting genetic variation, cellular function, and disease mechanisms. However, current human gene annotations are largely derived from transcriptomic...
www.biorxiv.org

Melé Lab
@melelab.bsky.social
· Mar 20

Melé Lab
@melelab.bsky.social
· Mar 20
Melé Lab
@melelab.bsky.social
· Mar 20

Melé Lab
@melelab.bsky.social
· Mar 20
Melé Lab
@melelab.bsky.social
· Mar 20
Melé Lab
@melelab.bsky.social
· Mar 20

Melé Lab
@melelab.bsky.social
· Mar 20
Melé Lab
@melelab.bsky.social
· Mar 20
Long-read transcriptomics of a diverse human cohort reveals widespread ancestry bias in gene annotation
Accurate gene annotations are fundamental for interpreting genetic variation, cellular function, and disease mechanisms. However, current human gene annotations are largely derived from transcriptomic...
www.biorxiv.org

