
Sergey Ovchinnikov
@sokrypton.org
3.8K followers
470 following
130 posts
Scientist, Assistant Professor at MIT biology, #FirstGen
Posts
Media
Videos
Starter Packs



Sergey Ovchinnikov
@sokrypton.org
· Sep 2
Reposted by Sergey Ovchinnikov

Reposted by Sergey Ovchinnikov

Sergey Ovchinnikov
@sokrypton.org
· Aug 6
Sergey Ovchinnikov
@sokrypton.org
· Aug 5
Reposted by Sergey Ovchinnikov


Sergey Ovchinnikov
@sokrypton.org
· Aug 5

How AlphaFold and related models predict protein-peptide complex structures
Protein-peptide interactions mediate many biological processes, and access to accurate structural models, through experimental determination or reliable computational prediction, is essential for unde...
www.biorxiv.org
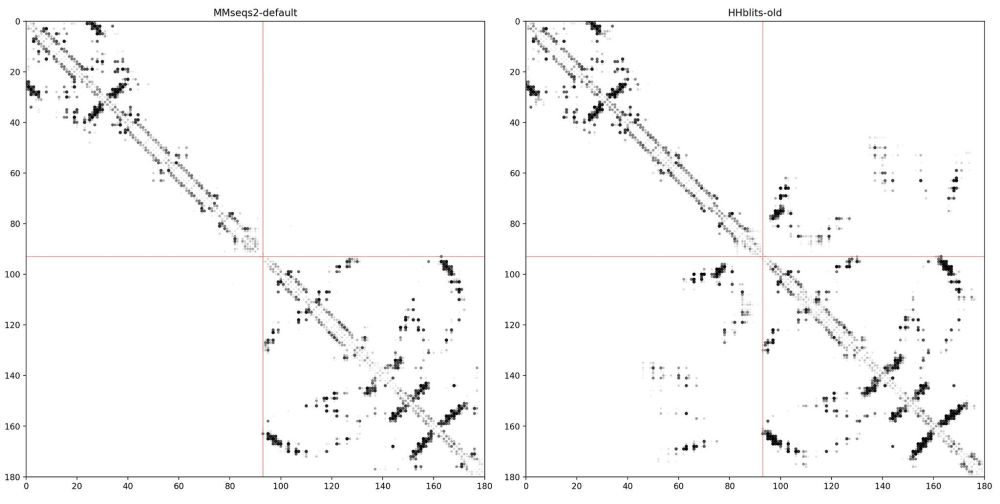
Sergey Ovchinnikov
@sokrypton.org
· Aug 5


Reposted by Sergey Ovchinnikov

Yo Akiyama
@yoakiyama.bsky.social
· Aug 5

Scaling down protein language modeling with MSA Pairformer
Recent efforts in protein language modeling have focused on scaling single-sequence models and their training data, requiring vast compute resources that limit accessibility. Although models that use ...
biorxiv.org
Sergey Ovchinnikov
@sokrypton.org
· Aug 3
Sergey Ovchinnikov
@sokrypton.org
· Aug 2
Sergey Ovchinnikov
@sokrypton.org
· Aug 2
Sergey Ovchinnikov
@sokrypton.org
· Aug 2
Sergey Ovchinnikov
@sokrypton.org
· Aug 2
Reposted by Sergey Ovchinnikov

Sergey Ovchinnikov
@sokrypton.org
· Aug 2
Sergey Ovchinnikov
@sokrypton.org
· Aug 2

Sergey Ovchinnikov
@sokrypton.org
· Jul 14