Yo Akiyama
@yoakiyama.bsky.social
730 followers
480 following
14 posts
MIT EECS PhD student in solab.org Building ML methods to understand and engineer biology
Posts
Media
Videos
Starter Packs



Yo Akiyama
@yoakiyama.bsky.social
· Aug 10
Reposted by Yo Akiyama

Reposted by Yo Akiyama











Yo Akiyama
@yoakiyama.bsky.social
· Aug 5
Yo Akiyama
@yoakiyama.bsky.social
· Aug 5

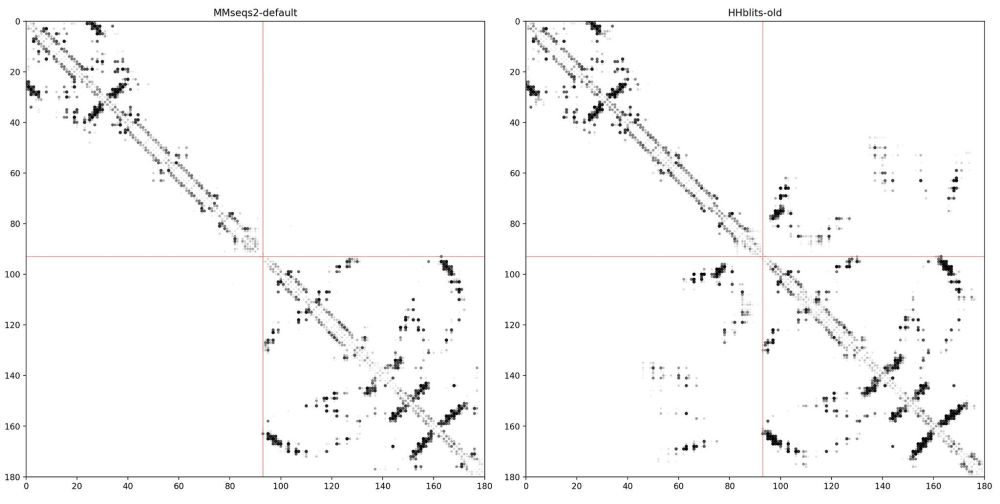
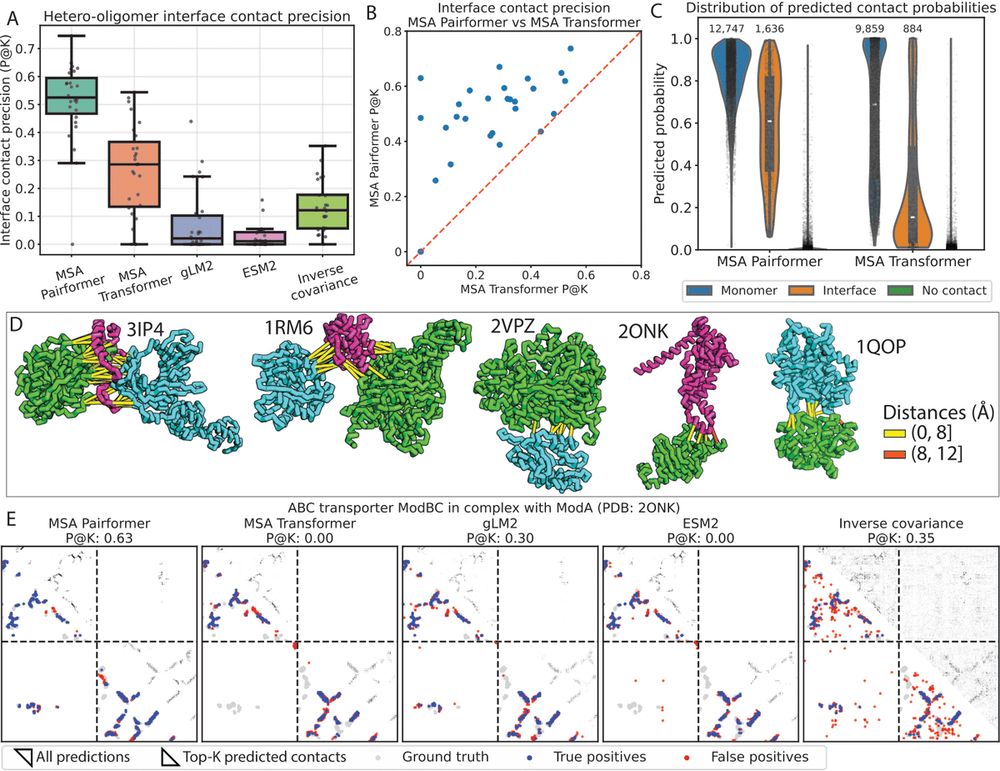
Scaling down protein language modeling with MSA Pairformer
Recent efforts in protein language modeling have focused on scaling single-sequence models and their training data, requiring vast compute resources that limit accessibility. Although models that use ...
biorxiv.org