Stephan Köstlbacher
@stephkoe.bsky.social
470 followers
310 following
32 posts
Former PostDoc at Wageningen University with Thijs Ettema
Studying ancient evolutionary transitions in prokaryotes using phylogenomics and structural modeling
Looking for next step in academic or translational research
he/him
Posts
Media
Videos
Starter Packs
Pinned

Stephan Köstlbacher
@stephkoe.bsky.social
· Jul 20

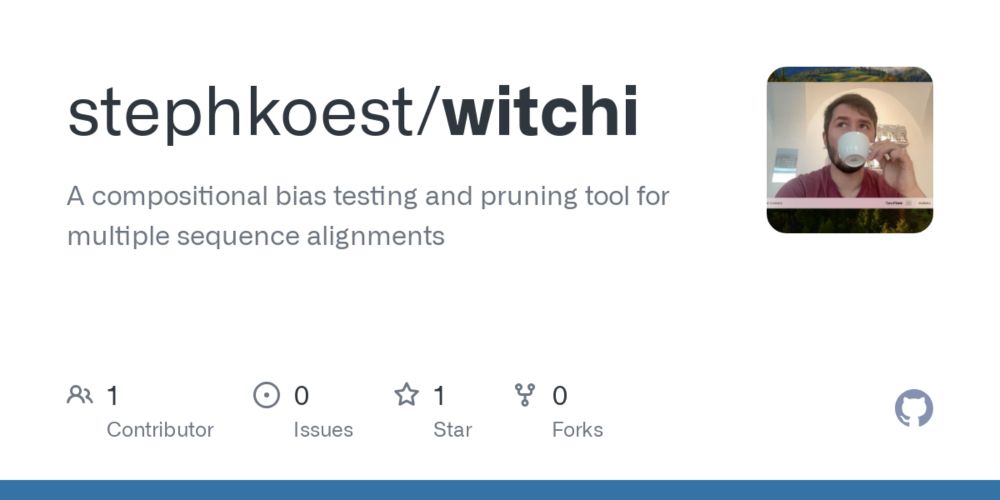
WitChi: Efficient Detection and Pruning of Compositional Bias in Phylogenomic Alignments Using Empirical Chi-Squared Testing
Convergent evolution, where unrelated taxa independently evolve similar nucleotide or amino acid compositions, can introduce compositional bias into biological sequence data. Such biases distort phylo...
www.biorxiv.org
Stephan Köstlbacher
@stephkoe.bsky.social
· Aug 22

Lorenzo Olivi
@loreoliv.bsky.social
· Aug 22

The Escherichia coli replication initiator DnaA is titrated on the chromosome - Nature Communications
A DNA-binding protein, DnaA, regulates the initiation of chromosomal replication in bacteria. Here, Olivi et al. show that the bacterial chromosome sequesters DnaA in a growth rate-dependent manner, s...
doi.org

Stephan Köstlbacher
@stephkoe.bsky.social
· Aug 22
Reposted by Stephan Köstlbacher

Stephan Köstlbacher
@stephkoe.bsky.social
· Jul 28
Stephan Köstlbacher
@stephkoe.bsky.social
· Jul 21
Reposted by Stephan Köstlbacher

Asaf Levy
@asaflevylab.bsky.social
· Jul 21

The Genetic Basis of Bacterial Adaptation to Hosts
Microbes colonize and interact with diverse multicellular hosts using specialized genes, many of which remain unidentified. Better understanding of host-associated gene functions is a key aspect of mi...
www.biorxiv.org
Stephan Köstlbacher
@stephkoe.bsky.social
· Jul 21
Stephan Köstlbacher
@stephkoe.bsky.social
· Jul 20
Stephan Köstlbacher
@stephkoe.bsky.social
· Jul 20
Reposted by Stephan Köstlbacher


Reposted by Stephan Köstlbacher

Stephan Köstlbacher
@stephkoe.bsky.social
· Jul 20





Stephan Köstlbacher
@stephkoe.bsky.social
· Jul 20

Stephan Köstlbacher
@stephkoe.bsky.social
· Jul 20
WitChi: Efficient Detection and Pruning of Compositional Bias in Phylogenomic Alignments Using Empirical Chi-Squared Testing
Convergent evolution, where unrelated taxa independently evolve similar nucleotide or amino acid compositions, can introduce compositional bias into biological sequence data. Such biases distort phylo...
www.biorxiv.org
Reposted by Stephan Köstlbacher

cemess_vienna
@cemess.bsky.social
· Jul 8
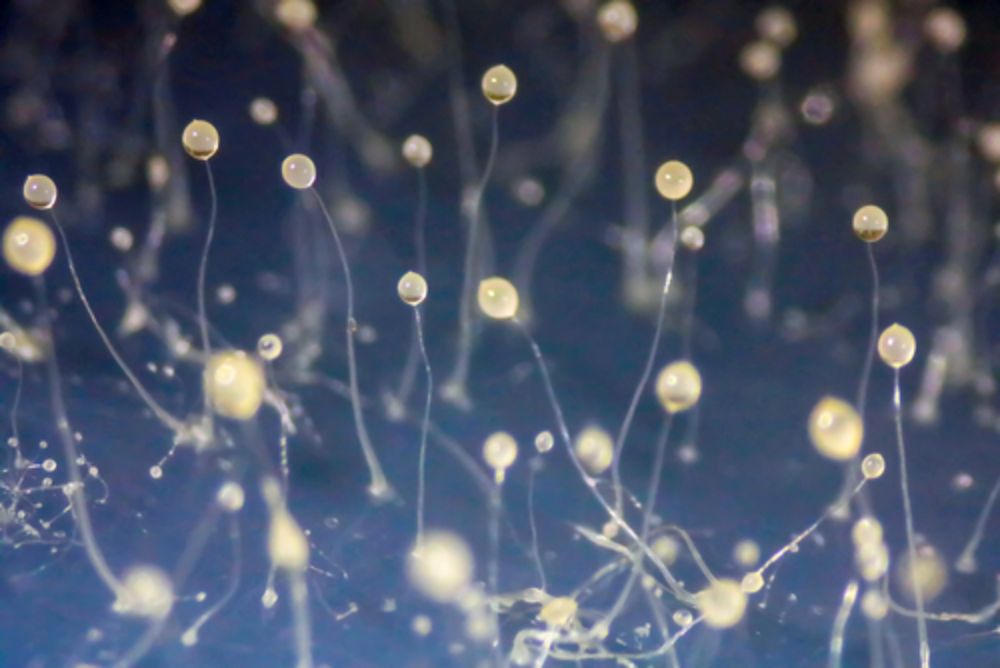
New Paper: Chlamydiae give up extracellular stage to thrive in social amoebae
Many intracellular bacteria rely on an extracellular stage to move between hosts – but what happens when that’s no longer needed? A new study, now published in Current Biology, reveals a striking exam...
cemess.univie.ac.at
Reposted by Stephan Köstlbacher

cemess_vienna
@cemess.bsky.social
· Jul 8
New Paper: Chlamydiae give up extracellular stage to thrive in social amoebae
Many intracellular bacteria rely on an extracellular stage to move between hosts – but what happens when that’s no longer needed? A new study, now published in Current Biology, reveals a striking exam...
cemess.univie.ac.at
Reposted by Stephan Köstlbacher