



Anthony Gitter
@anthonygitter.bsky.social
Computational biologist; Associate Prof. at University of Wisconsin-Madison; Jeanne M. Rowe Chair at Morgridge Institute
Our Assay2Mol manuscript was published at EMNLP 2025 doi.org/10.18653/v1/...
See the preprint thread below for a summary of the methodology, results, and code. We added more control experiments in this version related to protein sequence identity and generated molecule size.
See the preprint thread below for a summary of the methodology, results, and code. We added more control experiments in this version related to protein sequence identity and generated molecule size.
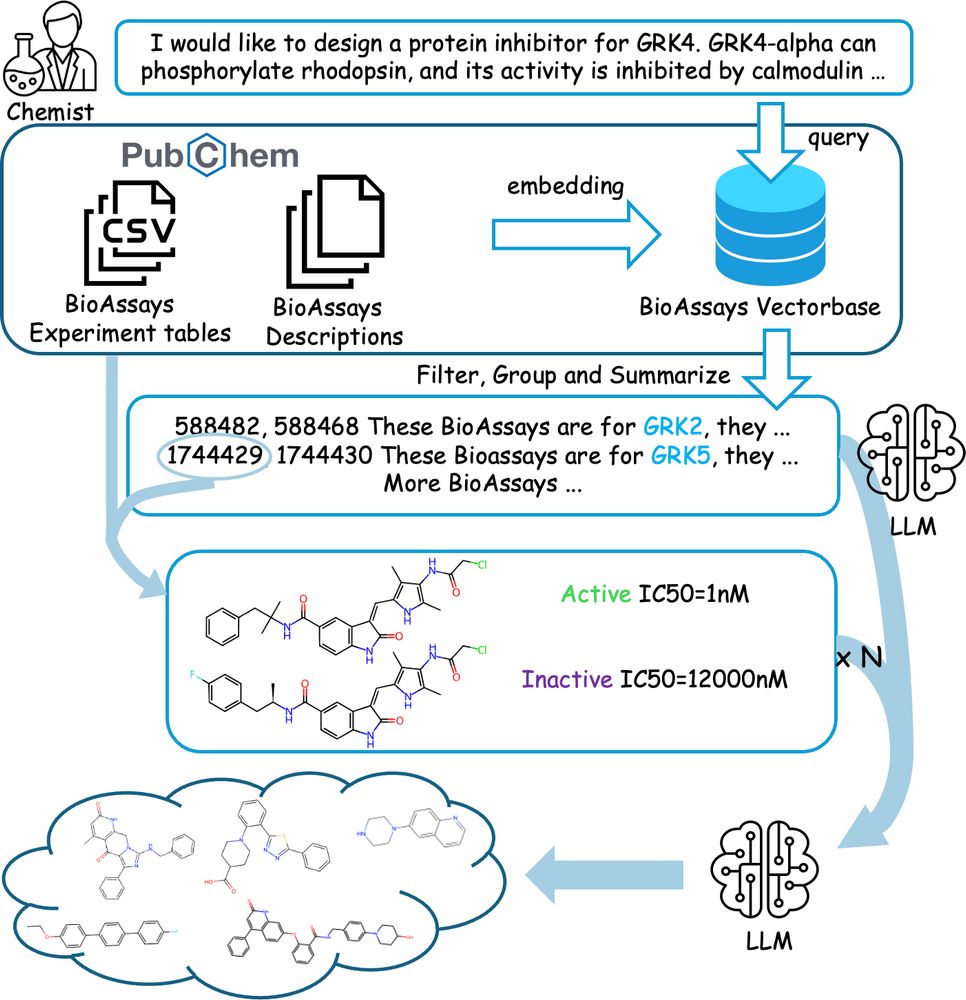
Our preprint Assay2Mol introduces uses PubChem chemical screening data as context when generating molecules with large language models. It uses assay descriptions and protocols to find relevant assays and that text plus active/inactive molecules as context for generation. 1/
November 21, 2025 at 3:19 PM
Our Assay2Mol manuscript was published at EMNLP 2025 doi.org/10.18653/v1/...
See the preprint thread below for a summary of the methodology, results, and code. We added more control experiments in this version related to protein sequence identity and generated molecule size.
See the preprint thread below for a summary of the methodology, results, and code. We added more control experiments in this version related to protein sequence identity and generated molecule size.
@hkws.bsky.social and I are creating the Madison AI for Proteins (MAIP) group to discuss early-stage research at monthly meetups, share computational resources, and grow this local community. Visit mad-ai-proteins.github.io to sign up for announcements and watch for our 2026 events.
November 20, 2025 at 4:29 PM
@hkws.bsky.social and I are creating the Madison AI for Proteins (MAIP) group to discuss early-stage research at monthly meetups, share computational resources, and grow this local community. Visit mad-ai-proteins.github.io to sign up for announcements and watch for our 2026 events.
Reposted by Anthony Gitter

This looks like a fantastic resource to study human kinase signalling. So much MS instrument time.

Chemical proteomics decrypts the kinases that shape the dynamic human phosphoproteome https://www.biorxiv.org/content/10.1101/2025.11.18.689017v1
November 19, 2025 at 6:18 AM
This looks like a fantastic resource to study human kinase signalling. So much MS instrument time.
Something fun and sciencey is coming soon to Madison
November 14, 2025 at 5:20 PM
Something fun and sciencey is coming soon to Madison
The journal version of our Multi-omic Pathway Analysis of Cells (MPAC) software is now out: doi.org/10.1093/bioi...
MPAC uses biological pathway graphs to model DNA copy number and gene expression changes and infer activity states of all pathway members.
MPAC uses biological pathway graphs to model DNA copy number and gene expression changes and infer activity states of all pathway members.

October 10, 2025 at 2:56 PM
The journal version of our Multi-omic Pathway Analysis of Cells (MPAC) software is now out: doi.org/10.1093/bioi...
MPAC uses biological pathway graphs to model DNA copy number and gene expression changes and infer activity states of all pathway members.
MPAC uses biological pathway graphs to model DNA copy number and gene expression changes and infer activity states of all pathway members.
Reposted by Anthony Gitter

AI + physics for protein engineering 🚀
Our collaboration with @anthonygitter.bsky.social is out in Nature Methods! We use synthetic data from molecular modeling to pretrain protein language models. Congrats to Sam Gelman and the team!
🔗 www.nature.com/articles/s41...
Our collaboration with @anthonygitter.bsky.social is out in Nature Methods! We use synthetic data from molecular modeling to pretrain protein language models. Congrats to Sam Gelman and the team!
🔗 www.nature.com/articles/s41...

Biophysics-based protein language models for protein engineering - Nature Methods
Mutational effect transfer learning (METL) is a protein language model framework that unites machine learning and biophysical modeling. Transformer-based neural networks are pretrained on biophysical simulation data to capture fundamental relationships between protein sequence, structure and energetics.
www.nature.com
October 1, 2025 at 7:07 PM
AI + physics for protein engineering 🚀
Our collaboration with @anthonygitter.bsky.social is out in Nature Methods! We use synthetic data from molecular modeling to pretrain protein language models. Congrats to Sam Gelman and the team!
🔗 www.nature.com/articles/s41...
Our collaboration with @anthonygitter.bsky.social is out in Nature Methods! We use synthetic data from molecular modeling to pretrain protein language models. Congrats to Sam Gelman and the team!
🔗 www.nature.com/articles/s41...
Reposted by Anthony Gitter

Does anyone know whether there's a functioning API to ESMfold?
(api.esmatlas.com/foldSequence... gives me Service Temporarily Unavailable)
(api.esmatlas.com/foldSequence... gives me Service Temporarily Unavailable)
September 30, 2025 at 2:11 PM
Does anyone know whether there's a functioning API to ESMfold?
(api.esmatlas.com/foldSequence... gives me Service Temporarily Unavailable)
(api.esmatlas.com/foldSequence... gives me Service Temporarily Unavailable)
The journal version of "Biophysics-based protein language models for protein engineering" with @philromero.bsky.social is live! Mutational Effect Transfer Learning (METL) is a protein language model trained on biophysical simulations that we use for protein engineering. 1/
doi.org/10.1038/s415...
doi.org/10.1038/s415...

Biophysics-based protein language models for protein engineering - Nature Methods
Mutational effect transfer learning (METL) is a protein language model framework that unites machine learning and biophysical modeling. Transformer-based neural networks are pretrained on biophysical ...
doi.org
September 11, 2025 at 5:00 PM
The journal version of "Biophysics-based protein language models for protein engineering" with @philromero.bsky.social is live! Mutational Effect Transfer Learning (METL) is a protein language model trained on biophysical simulations that we use for protein engineering. 1/
doi.org/10.1038/s415...
doi.org/10.1038/s415...
The journal version of our paper 'Chemical Language Model Linker: Blending Text and Molecules with Modular Adapters' is out doi.org/10.1021/acs....
ChemLML is a method for text-based conditional molecule generation that uses pretrained text models like SciBERT, Galactica, or T5.
ChemLML is a method for text-based conditional molecule generation that uses pretrained text models like SciBERT, Galactica, or T5.

Chemical Language Model Linker: Blending Text and Molecules with Modular Adapters
The development of large language models and multimodal models has enabled the appealing idea of generating novel molecules from text descriptions. Generative modeling would shift the paradigm from relying on large-scale chemical screening to find molecules with desired properties to directly generating those molecules. However, multimodal models combining text and molecules are often trained from scratch, without leveraging existing high-quality pretrained models. Training from scratch consumes more computational resources and prohibits model scaling. In contrast, we propose a lightweight adapter-based strategy named Chemical Language Model Linker (ChemLML). ChemLML blends the two single domain models and obtains conditional molecular generation from text descriptions while still operating in the specialized embedding spaces of the molecular domain. ChemLML can tailor diverse pretrained text models for molecule generation by training relatively few adapter parameters. We find that the choice of molecular representation used within ChemLML, SMILES versus SELFIES, has a strong influence on conditional molecular generation performance. SMILES is often preferable despite not guaranteeing valid molecules. We raise issues in using the entire PubChem data set of molecules and their associated descriptions for evaluating molecule generation and provide a filtered version of the data set as a generation test set. To demonstrate how ChemLML could be used in practice, we generate candidate protein inhibitors and use docking to assess their quality and also generate candidate membrane permeable molecules.
doi.org
August 22, 2025 at 1:36 PM
The journal version of our paper 'Chemical Language Model Linker: Blending Text and Molecules with Modular Adapters' is out doi.org/10.1021/acs....
ChemLML is a method for text-based conditional molecule generation that uses pretrained text models like SciBERT, Galactica, or T5.
ChemLML is a method for text-based conditional molecule generation that uses pretrained text models like SciBERT, Galactica, or T5.
Reposted by Anthony Gitter

🚨New paper 🚨
Can protein language models help us fight viral outbreaks? Not yet. Here’s why 🧵👇
1/12
Can protein language models help us fight viral outbreaks? Not yet. Here’s why 🧵👇
1/12

August 17, 2025 at 3:42 AM
🚨New paper 🚨
Can protein language models help us fight viral outbreaks? Not yet. Here’s why 🧵👇
1/12
Can protein language models help us fight viral outbreaks? Not yet. Here’s why 🧵👇
1/12
Our preprint Assay2Mol introduces uses PubChem chemical screening data as context when generating molecules with large language models. It uses assay descriptions and protocols to find relevant assays and that text plus active/inactive molecules as context for generation. 1/
July 18, 2025 at 3:13 PM
Our preprint Assay2Mol introduces uses PubChem chemical screening data as context when generating molecules with large language models. It uses assay descriptions and protocols to find relevant assays and that text plus active/inactive molecules as context for generation. 1/
Reposted by Anthony Gitter

Nobody is commenting on this little nugget from Fig 1?

July 18, 2025 at 2:55 PM
Nobody is commenting on this little nugget from Fig 1?
Reposted by Anthony Gitter

Happy to share this interview with Weijie Zhao from NSR at #OxfordUniversityPress. It covers questions I’m often asked—why I chose Korea, AlphaFold2, my unconventional journey into academia, and research insights. Thanks again for the fun conversation.
📄 academic.oup.com/nsr/article/...
📄 academic.oup.com/nsr/article/...

New methods are revolutionizing biology: an interview with Martin Steinegger
Martin Steinegger, who is the only non-DeepMind-affiliated author of the AlphaFold2 Nature paper, offers unique insights and personal reflections.
academic.oup.com
May 19, 2025 at 12:08 PM
Happy to share this interview with Weijie Zhao from NSR at #OxfordUniversityPress. It covers questions I’m often asked—why I chose Korea, AlphaFold2, my unconventional journey into academia, and research insights. Thanks again for the fun conversation.
📄 academic.oup.com/nsr/article/...
📄 academic.oup.com/nsr/article/...
Reposted by Anthony Gitter

To honor the 75th anniversary of @NSF, RCSB PDB Intern Xinyi Christine Zhang created posters to celebrate the science made possible by the NSF and RCSB PDB.
Explore these images and learn how protein research is changing our world. #NSFfunded #NSF75
pdb101.rcsb.org/lear...
Explore these images and learn how protein research is changing our world. #NSFfunded #NSF75
pdb101.rcsb.org/lear...
PDB101: Learn: Other Resources: Commemorating 75 Years of Discovery and Innovation at the NSF
Download images celebrating NSF and PDB milestones
pdb101.rcsb.org
May 8, 2025 at 4:18 PM
To honor the 75th anniversary of @NSF, RCSB PDB Intern Xinyi Christine Zhang created posters to celebrate the science made possible by the NSF and RCSB PDB.
Explore these images and learn how protein research is changing our world. #NSFfunded #NSF75
pdb101.rcsb.org/lear...
Explore these images and learn how protein research is changing our world. #NSFfunded #NSF75
pdb101.rcsb.org/lear...
My first post is a niche and personal shout out to @michaelhoffman.bsky.social, the person who asked me most often if I am on Bluesky yet.
April 3, 2025 at 11:23 PM
My first post is a niche and personal shout out to @michaelhoffman.bsky.social, the person who asked me most often if I am on Bluesky yet.