



Mingxun Wang
@mingxunwang.bsky.social
Assistant Professor @ UCR
Computational Mass Spectrometry, Bioinformatics.
#massspec #molecularnetworking #GNPS #MassQL
https://www.cs.ucr.edu/~mingxunw/
Computational Mass Spectrometry, Bioinformatics.
#massspec #molecularnetworking #GNPS #MassQL
https://www.cs.ucr.edu/~mingxunw/
Pinned
Mingxun Wang
@mingxunwang.bsky.social
· May 12
I am thrilled to share after years of work/procrastination that the MassQL manuscript is finally published in @natmethods.nature.com - "A universal language for finding mass spectrometry data patterns". This was an team effort from all co-authors that helped shape MassQL and how it could be used.
We are hiring for a metabolomics position here at UC Riverside! Come join the vibrant research and mass spectrometry community here!
aprecruit.ucr.edu/JPF02151
aprecruit.ucr.edu/JPF02151

Assistant Professor in Computational and/or Analytical Metabolomics
University of California, Riverside is hiring. Apply now!
aprecruit.ucr.edu
November 19, 2025 at 11:52 PM
We are hiring for a metabolomics position here at UC Riverside! Come join the vibrant research and mass spectrometry community here!
aprecruit.ucr.edu/JPF02151
aprecruit.ucr.edu/JPF02151
To help make more mass spec data accessible - we've just rolled out a change to enable universal spectrum identifier resolution and plotting directly from mzML files in Zenodo. We're growing support from more sources in GNPS2 for public data reanalysis!
metabolomics-usi.gnps2.org/dashinterfac...
metabolomics-usi.gnps2.org/dashinterfac...

September 9, 2025 at 10:34 PM
To help make more mass spec data accessible - we've just rolled out a change to enable universal spectrum identifier resolution and plotting directly from mzML files in Zenodo. We're growing support from more sources in GNPS2 for public data reanalysis!
metabolomics-usi.gnps2.org/dashinterfac...
metabolomics-usi.gnps2.org/dashinterfac...
Reposted by Mingxun Wang

Interested in a co-authorship?
We’re building a tool for repository-scale untargeted #metabolomics and #exposomics of #environmental data. To make it the best it can be, we’re looking for people willing to share high-resolution LC-MS/MS (DDA) data from #water, #soil, #sediment, and related samples.
We’re building a tool for repository-scale untargeted #metabolomics and #exposomics of #environmental data. To make it the best it can be, we’re looking for people willing to share high-resolution LC-MS/MS (DDA) data from #water, #soil, #sediment, and related samples.

August 26, 2025 at 7:51 PM
Interested in a co-authorship?
We’re building a tool for repository-scale untargeted #metabolomics and #exposomics of #environmental data. To make it the best it can be, we’re looking for people willing to share high-resolution LC-MS/MS (DDA) data from #water, #soil, #sediment, and related samples.
We’re building a tool for repository-scale untargeted #metabolomics and #exposomics of #environmental data. To make it the best it can be, we’re looking for people willing to share high-resolution LC-MS/MS (DDA) data from #water, #soil, #sediment, and related samples.
GNPS2 and associated services will be down for power maintenance tonight and into tomorrow.
August 2, 2025 at 4:48 AM
GNPS2 and associated services will be down for power maintenance tonight and into tomorrow.
Reposted by Mingxun Wang
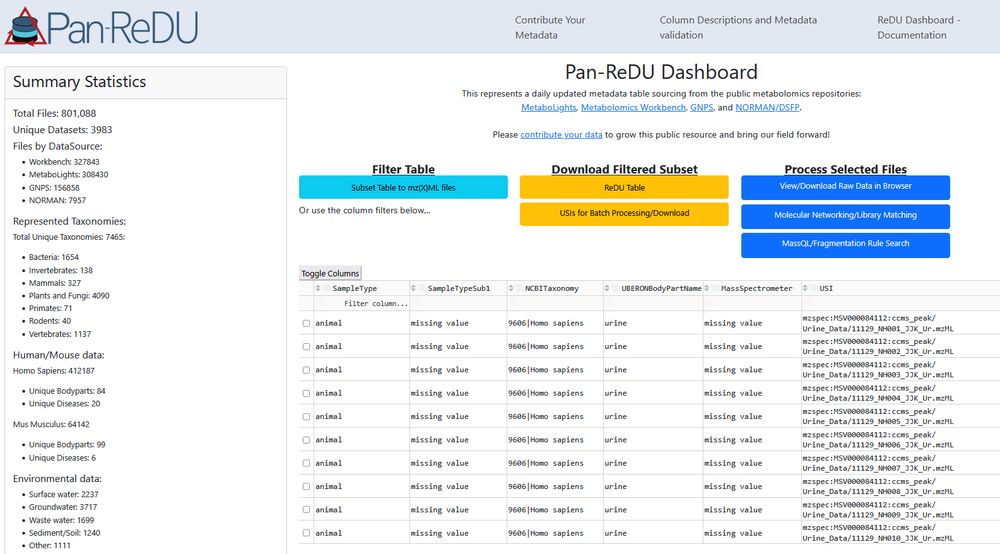
We just crossed the 800,000 files mark in Pan-ReDU. That's 800,000 public #metabolomics raw data files with harmonized metadata that can be re-analyzed to learn about new molecules and bio-distributions. 🎉 redu.gnps2.org
July 9, 2025 at 12:55 AM
We just crossed the 800,000 files mark in Pan-ReDU. That's 800,000 public #metabolomics raw data files with harmonized metadata that can be re-analyzed to learn about new molecules and bio-distributions. 🎉 redu.gnps2.org
Reposted by Mingxun Wang

The Mass Spectrometry Query Language (MassQL) is an open-source language for instrument-independent searching across mass spectrometry data for complex patterns of interest via concise and expressive queries without the need for programming skills.
www.nature.com/articles/s41...
www.nature.com/articles/s41...

May 13, 2025 at 3:25 PM
The Mass Spectrometry Query Language (MassQL) is an open-source language for instrument-independent searching across mass spectrometry data for complex patterns of interest via concise and expressive queries without the need for programming skills.
www.nature.com/articles/s41...
www.nature.com/articles/s41...
May 12, 2025 at 10:27 PM
I am thrilled to share after years of work/procrastination that the MassQL manuscript is finally published in @natmethods.nature.com - "A universal language for finding mass spectrometry data patterns". This was an team effort from all co-authors that helped shape MassQL and how it could be used.
May 12, 2025 at 6:10 PM
I am thrilled to share after years of work/procrastination that the MassQL manuscript is finally published in @natmethods.nature.com - "A universal language for finding mass spectrometry data patterns". This was an team effort from all co-authors that helped shape MassQL and how it could be used.
Reposted by Mingxun Wang

We are back online!
GNPS2 is planning on being down for server maintenance tomorrow at 12PM PST. We expect 5 hours of downtime to move servers, bring online new storage, and increase networking performance.
March 6, 2025 at 4:42 AM
We are back online!
Reposted by Mingxun Wang

MS-RT: A Method for Evaluating MS/MS Clustering Performance for Metabolomics Data pubs.acs.org/doi/10.1021/...

MS-RT: A Method for Evaluating MS/MS Clustering Performance for Metabolomics Data
The clustering of tandem mass spectra (MS/MS) is a crucial computational step to deduplicate repeated acquisitions in data-dependent experiments. This technique is essential in untargeted metabolomics, particularly with high-throughput mass spectrometers capable of generating hundreds of MS/MS spectra per second. Despite advancements in MS/MS clustering algorithms in proteomics, their performance in metabolomics has not been extensively evaluated due to the lack of database search tools with false discovery rate control for molecule identification. To bridge this gap, this study introduces the MS1-retention time (MS-RT) method to assess MS/MS clustering performance in metabolomics data sets. Here, we validate MS-RT by comparing MS-RT to established proteomics clustering evaluation approaches that utilize database search identifications. Additionally, we evaluate the performance of several MS/MS clustering tools on metabolomics data sets, highlighting their advantages and drawbacks. This MS-RT method and the MS/MS clustering tool benchmarking will provide valuable real world practical recommendations for tools and set the stage for future advancements in metabolomics MS/MS clustering.
pubs.acs.org
March 6, 2025 at 8:34 PM
MS-RT: A Method for Evaluating MS/MS Clustering Performance for Metabolomics Data pubs.acs.org/doi/10.1021/...
Reposted by Mingxun Wang

Congrats to all the people who put in tremendous effort to make this study possible. Such a fun project!
www.cell.com/cell/fulltex...
#space #metabolomics #microbes
www.cell.com/cell/fulltex...
#space #metabolomics #microbes
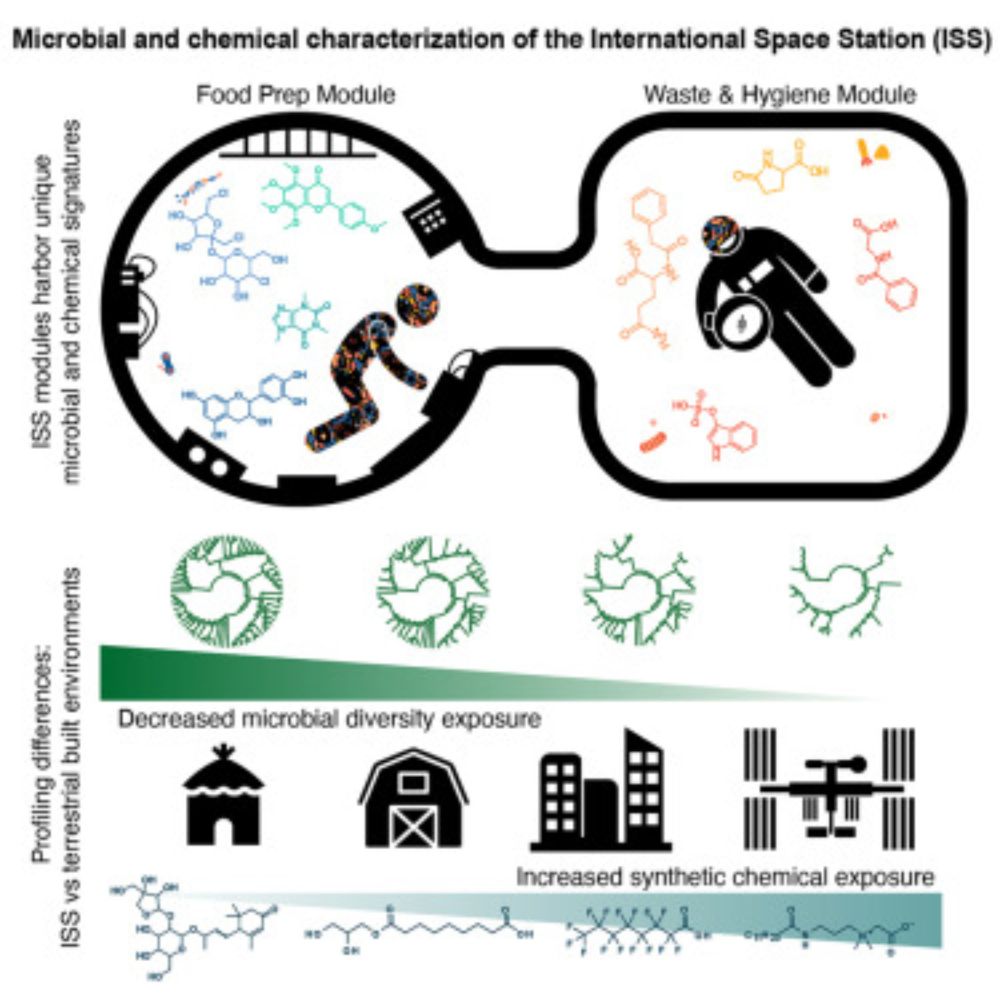
The International Space Station has a unique and extreme microbial and chemical environment driven by use patterns
With long-term space travel and extraterrestrial habitation becoming feasible, understanding
how space environmental exposures, microbial communities, and molecular profiles differ
from Earth is cruci...
www.cell.com
March 6, 2025 at 5:40 PM
Congrats to all the people who put in tremendous effort to make this study possible. Such a fun project!
www.cell.com/cell/fulltex...
#space #metabolomics #microbes
www.cell.com/cell/fulltex...
#space #metabolomics #microbes
Reposted by Mingxun Wang

#FeaturedProtocol this week is a #reversemetabolomics protocol, in which a tandem #massspec spectrum is used as a search term to probe public #metabolomic data, enabling discovery of new metabolic associations bit.ly/4hdyQQF

A guide to reverse metabolomics—a framework for big data discovery strategy - Nature Protocols
In this reverse metabolomics protocol, a tandem mass spectrometry spectrum is used as a search term to probe public metabolomic data. Analysis of the metadata connected with these search results enabl...
bit.ly
March 4, 2025 at 3:21 PM
#FeaturedProtocol this week is a #reversemetabolomics protocol, in which a tandem #massspec spectrum is used as a search term to probe public #metabolomic data, enabling discovery of new metabolic associations bit.ly/4hdyQQF
I am excited to share this new paper out in JPR - "MS-RT: A Method for Evaluating MS/MS Clustering Performance for Metabolomics Data." This work introduces the MS-RT method to assess MS/MS clustering accuracy on metabolomics data.
doi.org/10.1021/acs....
doi.org/10.1021/acs....
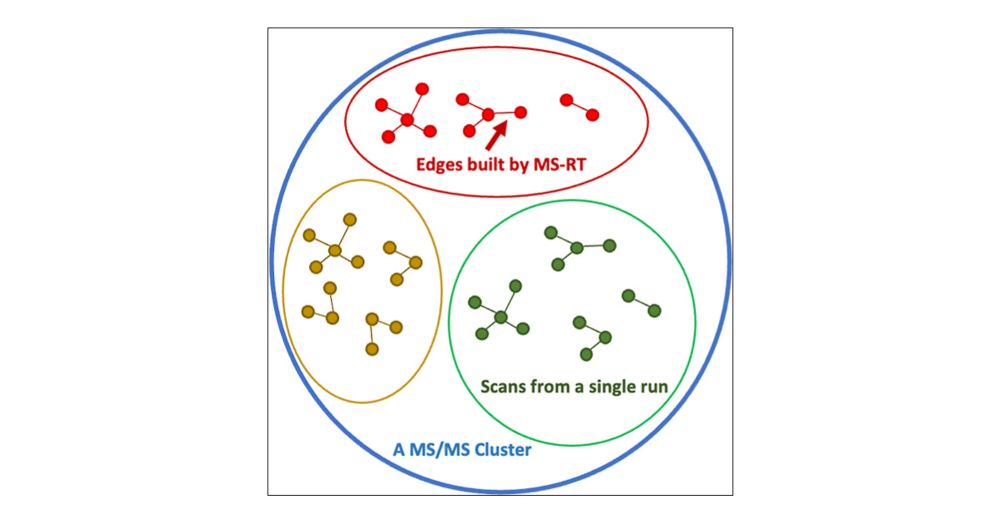
MS-RT: A Method for Evaluating MS/MS Clustering Performance for Metabolomics Data
The clustering of tandem mass spectra (MS/MS) is a crucial computational step to deduplicate repeated acquisitions in data-dependent experiments. This technique is essential in untargeted metabolomics, particularly with high-throughput mass spectrometers capable of generating hundreds of MS/MS spectra per second. Despite advancements in MS/MS clustering algorithms in proteomics, their performance in metabolomics has not been extensively evaluated due to the lack of database search tools with false discovery rate control for molecule identification. To bridge this gap, this study introduces the MS1-retention time (MS-RT) method to assess MS/MS clustering performance in metabolomics data sets. Here, we validate MS-RT by comparing MS-RT to established proteomics clustering evaluation approaches that utilize database search identifications. Additionally, we evaluate the performance of several MS/MS clustering tools on metabolomics data sets, highlighting their advantages and drawbacks. This MS-RT method and the MS/MS clustering tool benchmarking will provide valuable real world practical recommendations for tools and set the stage for future advancements in metabolomics MS/MS clustering.
doi.org
March 5, 2025 at 5:34 PM
I am excited to share this new paper out in JPR - "MS-RT: A Method for Evaluating MS/MS Clustering Performance for Metabolomics Data." This work introduces the MS-RT method to assess MS/MS clustering accuracy on metabolomics data.
doi.org/10.1021/acs....
doi.org/10.1021/acs....
Reposted by Mingxun Wang
GNPS2 is planning on being down for server maintenance tomorrow at 12PM PST. We expect 5 hours of downtime to move servers, bring online new storage, and increase networking performance.
March 5, 2025 at 3:45 AM
GNPS2 is planning on being down for server maintenance tomorrow at 12PM PST. We expect 5 hours of downtime to move servers, bring online new storage, and increase networking performance.
We are back online!
GNPS2 will be down for the next hour for a new release update!
January 28, 2025 at 11:19 PM
We are back online!
GNPS2 will be down for the next hour for a new release update!
January 28, 2025 at 8:13 PM
GNPS2 will be down for the next hour for a new release update!
Reposted by Mingxun Wang

🚨 Webinar Reminder! 🚨
Don’t forget to join us for our first webinar of the year:
“Learning From Repository-Scale Untargeted Metabolomics Data”
📅 Date: 22 January
🕒 Time: 3 PM UTC
#MetabolomicsSociety #MetSoc #Metabolomics #ECR #TeamMassSpec #EMNMetSoc
Don’t forget to join us for our first webinar of the year:
“Learning From Repository-Scale Untargeted Metabolomics Data”
📅 Date: 22 January
🕒 Time: 3 PM UTC
#MetabolomicsSociety #MetSoc #Metabolomics #ECR #TeamMassSpec #EMNMetSoc
🎉 First webinar for 2025!
On 22 January at 3pm UTC, we will host Dr Wout Bittremieux who will deliver a talk on an approach to untargeted metabolomics data annotation!
✒️ Register here for free: tinyurl.com/2v654vp9
We look forward to seeing you there 👋
On 22 January at 3pm UTC, we will host Dr Wout Bittremieux who will deliver a talk on an approach to untargeted metabolomics data annotation!
✒️ Register here for free: tinyurl.com/2v654vp9
We look forward to seeing you there 👋

January 17, 2025 at 1:05 AM
🚨 Webinar Reminder! 🚨
Don’t forget to join us for our first webinar of the year:
“Learning From Repository-Scale Untargeted Metabolomics Data”
📅 Date: 22 January
🕒 Time: 3 PM UTC
#MetabolomicsSociety #MetSoc #Metabolomics #ECR #TeamMassSpec #EMNMetSoc
Don’t forget to join us for our first webinar of the year:
“Learning From Repository-Scale Untargeted Metabolomics Data”
📅 Date: 22 January
🕒 Time: 3 PM UTC
#MetabolomicsSociety #MetSoc #Metabolomics #ECR #TeamMassSpec #EMNMetSoc
It was a wonderful working with so many old friends again at #SIMB. Thank you to all the participants who were super engaged and asked amazing questions. This kind of feedback is what makes building tools so worthwhile for the community and improves future developments.
Such a wonderful SIMB workshop on @gnps2.bsky.social covering classical mol networking, FBMN, ion identity based mol networking, FBMN-STATS, gnps-dashboard, CMMC-kb, Modifinder, MicrobeMASST, MassQL, and limited ReDU - many were covered at a workshop for the first time. 1/4




January 10, 2025 at 6:07 PM
It was a wonderful working with so many old friends again at #SIMB. Thank you to all the participants who were super engaged and asked amazing questions. This kind of feedback is what makes building tools so worthwhile for the community and improves future developments.
We are back!
GNPS2 will be in maintenance this morning with some electricity upgrades in our data center room. We anticipate GNPS2 will come back online in a few hours.
December 18, 2024 at 5:36 PM
We are back!
Reposted by Mingxun Wang
GNPS2 will be in maintenance this morning with some electricity upgrades in our data center room. We anticipate GNPS2 will come back online in a few hours.
December 18, 2024 at 4:06 PM
GNPS2 will be in maintenance this morning with some electricity upgrades in our data center room. We anticipate GNPS2 will come back online in a few hours.
Reposted by Mingxun Wang

My department is hiring a fungal biologist! aprecruit.ucr.edu/JPF01990

Assistant Professor in Fungal Biology
University of California, Riverside is hiring. Apply now!
aprecruit.ucr.edu
December 10, 2024 at 7:14 PM
My department is hiring a fungal biologist! aprecruit.ucr.edu/JPF01990
Reposted by Mingxun Wang

Remember to VOTE in the ASMS Board election. All current members started to receive online ballot via email on Dec 2. Polls close on December 19.
Learn about the election & meet the candidates here:
www.asms.org/about/board-...
Learn about the election & meet the candidates here:
www.asms.org/about/board-...

December 4, 2024 at 4:36 PM
Remember to VOTE in the ASMS Board election. All current members started to receive online ballot via email on Dec 2. Polls close on December 19.
Learn about the election & meet the candidates here:
www.asms.org/about/board-...
Learn about the election & meet the candidates here:
www.asms.org/about/board-...
Reposted by Mingxun Wang

December 4, 2024 at 12:42 AM
Reposted by Mingxun Wang

To register docs.google.com/forms/d/e/1F...
December 3, 2024 at 8:17 PM
To register docs.google.com/forms/d/e/1F...
Reposted by Mingxun Wang
The CMMC is hosting a reverse metabolomics workshop. The foundation of these papers www.nature.com/articles/s41..., www.cell.com/cell/fulltex... and www.biorxiv.org/content/10.1... - here searched @gnps2.bsky.social only now @metabolights.bsky.social and @metabolomics workbench as well. #metabolome

December 3, 2024 at 8:16 PM
The CMMC is hosting a reverse metabolomics workshop. The foundation of these papers www.nature.com/articles/s41..., www.cell.com/cell/fulltex... and www.biorxiv.org/content/10.1... - here searched @gnps2.bsky.social only now @metabolights.bsky.social and @metabolomics workbench as well. #metabolome