



Marco Molari
@mmolari.bsky.social
PostDoc @ Biozentrum - Basel in the group of Richard Neher
Interested in microbial genomics, pangenome graphs & evolution 🧬🦠💻
Interested in microbial genomics, pangenome graphs & evolution 🧬🦠💻
Reposted by Marco Molari

I want to spell this out in case the implications aren't clear:
This means all public tools/webapps of GISAID data (all the ones you've been used to seeing thru the pandemic, as far as we can tell) are prohibited.
The file allowed this. Cut that - cut off all tools the public & others were using.
This means all public tools/webapps of GISAID data (all the ones you've been used to seeing thru the pandemic, as far as we can tell) are prohibited.
The file allowed this. Cut that - cut off all tools the public & others were using.

On Oct 1, 2025, GISAID informed us that they had ended updates to the flat file of SARS-CoV-2 genomic sequences and associated metadata that we had used to update Nextstrain analyses since Feb 2020. GISAID's stated rationale was that their "resources are limited". 1/5

November 7, 2025 at 2:41 PM
I want to spell this out in case the implications aren't clear:
This means all public tools/webapps of GISAID data (all the ones you've been used to seeing thru the pandemic, as far as we can tell) are prohibited.
The file allowed this. Cut that - cut off all tools the public & others were using.
This means all public tools/webapps of GISAID data (all the ones you've been used to seeing thru the pandemic, as far as we can tell) are prohibited.
The file allowed this. Cut that - cut off all tools the public & others were using.
Reposted by Marco Molari

Delighted to see our paper studying the evolution of plasmids over the last 100 years, now out! Years of work by Adrian Cazares, also Nick Thomson @sangerinstitute.bsky.social - this version much improved over the preprint. Final version should be open access, apols.
Thread 1/n
Thread 1/n

September 25, 2025 at 9:29 PM
Delighted to see our paper studying the evolution of plasmids over the last 100 years, now out! Years of work by Adrian Cazares, also Nick Thomson @sangerinstitute.bsky.social - this version much improved over the preprint. Final version should be open access, apols.
Thread 1/n
Thread 1/n
Reposted by Marco Molari

We're looking for highly motivated candidates for PhD positions in the physics of living systems.
Our group uses theoretical physics to understand how cells collectively self-organize.
If you're interested, get in touch & check out the Biozentrum PhD Fellowship program, deadline October 12th!
Our group uses theoretical physics to understand how cells collectively self-organize.
If you're interested, get in touch & check out the Biozentrum PhD Fellowship program, deadline October 12th!

Apply now for the prestigious and independent #BiozentrumPhDFellowships. Great science. Unique rotation-based selection of research group and other incentives. The summer call is open until October 12, 2025. bit.ly/4caiqqX @biozentrum.unibas.ch @unibas.ch #fellowship #PhD#Switzerland

Biozentrum PhD Fellowships
Share your passion for life sciences. If you are talented and highly motivated, want to broaden your horizons and are interested in a wide range of research topics, apply for one of the sought after B...
bit.ly
September 15, 2025 at 10:04 AM
We're looking for highly motivated candidates for PhD positions in the physics of living systems.
Our group uses theoretical physics to understand how cells collectively self-organize.
If you're interested, get in touch & check out the Biozentrum PhD Fellowship program, deadline October 12th!
Our group uses theoretical physics to understand how cells collectively self-organize.
If you're interested, get in touch & check out the Biozentrum PhD Fellowship program, deadline October 12th!
Reposted by Marco Molari

Willing to join us @pasteur.fr for a PhD for a project on how interactions between mobile genetic elements shape bacterial adaptation? Subject to be tailored to candidates with keen interest in evolution, genomics, computational biology, microbiology. Check www.pasteur.fr/en/education...
September 8, 2025 at 8:56 AM
Willing to join us @pasteur.fr for a PhD for a project on how interactions between mobile genetic elements shape bacterial adaptation? Subject to be tailored to candidates with keen interest in evolution, genomics, computational biology, microbiology. Check www.pasteur.fr/en/education...
Reposted by Marco Molari
It was a great pleasure to contribute to this work by Jemma Fendley, @mmolari.bsky.social, and Boris Shraiman on pan-genomes, linkage, and recombination in phage genomes.
We analyzed data collected by the fantastic SEA-PHAGES program in phagesdb.org.
[1/6]
www.biorxiv.org/content/10.1...
We analyzed data collected by the fantastic SEA-PHAGES program in phagesdb.org.
[1/6]
www.biorxiv.org/content/10.1...
The Actinobacteriophage Database | Home
phagesdb.org
August 23, 2025 at 4:21 PM
It was a great pleasure to contribute to this work by Jemma Fendley, @mmolari.bsky.social, and Boris Shraiman on pan-genomes, linkage, and recombination in phage genomes.
We analyzed data collected by the fantastic SEA-PHAGES program in phagesdb.org.
[1/6]
www.biorxiv.org/content/10.1...
We analyzed data collected by the fantastic SEA-PHAGES program in phagesdb.org.
[1/6]
www.biorxiv.org/content/10.1...
Reposted by Marco Molari
📢New job posting!📢
Are you excited by the idea of building global infrastructure to make pathogen sequencing more accessible, interpretable, and equitable? 🧑🏻💻🧬
My group at @swisstph.ch has an opening working with ARTIC2, @pathoplexus.org, & Loculus - read on!
1/5
Are you excited by the idea of building global infrastructure to make pathogen sequencing more accessible, interpretable, and equitable? 🧑🏻💻🧬
My group at @swisstph.ch has an opening working with ARTIC2, @pathoplexus.org, & Loculus - read on!
1/5

July 25, 2025 at 10:44 AM
📢New job posting!📢
Are you excited by the idea of building global infrastructure to make pathogen sequencing more accessible, interpretable, and equitable? 🧑🏻💻🧬
My group at @swisstph.ch has an opening working with ARTIC2, @pathoplexus.org, & Loculus - read on!
1/5
Are you excited by the idea of building global infrastructure to make pathogen sequencing more accessible, interpretable, and equitable? 🧑🏻💻🧬
My group at @swisstph.ch has an opening working with ARTIC2, @pathoplexus.org, & Loculus - read on!
1/5
Reposted by Marco Molari

Bonsai is out! Finally, we can visually explore high-dimensional data in a distortion-free way. Try it out! It works from scRNA-seq data (bonsai.unibas.ch/bonsai-scout...) up to football statistics (bonsai.unibas.ch/bonsai-scout...).
Ever wondered why low-dimensional embeddings like t-SNE
May 9, 2025 at 11:55 AM
Bonsai is out! Finally, we can visually explore high-dimensional data in a distortion-free way. Try it out! It works from scRNA-seq data (bonsai.unibas.ch/bonsai-scout...) up to football statistics (bonsai.unibas.ch/bonsai-scout...).
Ever wondered why low-dimensional embeddings like t-SNE
Reposted by Marco Molari

Agencies within the Trump administration have flagged hundreds of words to limit or avoid, according to a compilation of government documents. These terms appeared in government memos, in official and unofficial agency guidance and in other documents. www.nytimes.com/interactive/...

March 10, 2025 at 12:25 AM
Agencies within the Trump administration have flagged hundreds of words to limit or avoid, according to a compilation of government documents. These terms appeared in government memos, in official and unofficial agency guidance and in other documents. www.nytimes.com/interactive/...
Reposted by Marco Molari
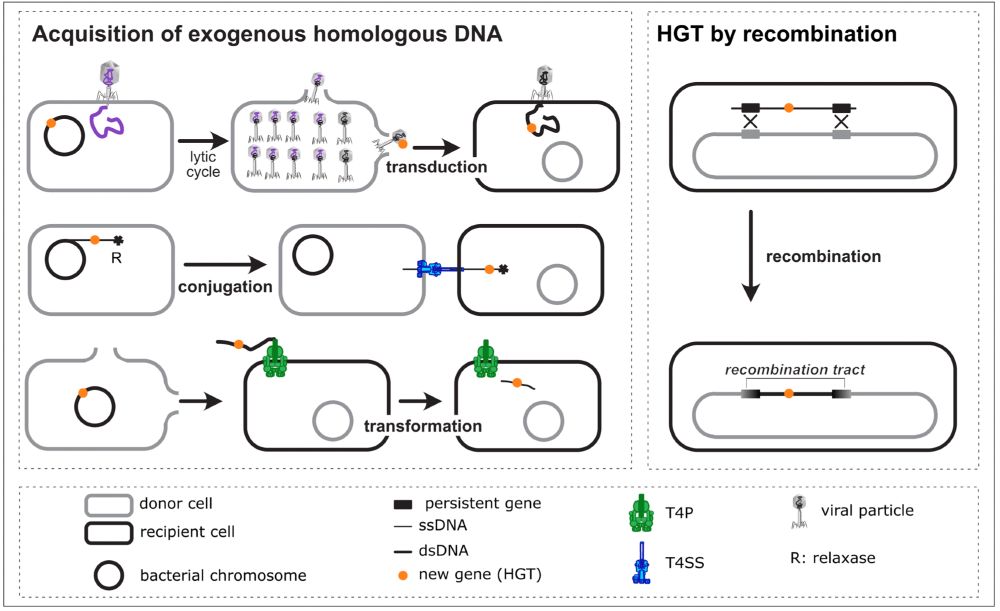
The contribution of natural transformation for the acquisition of novel genes has been notoriously difficult to quantify because it relies on recombination (which is affected by other processes). Here's a first estimate : doi.org/10.1101/2025... (for the very busy: 1-6% of gene gains) #MicroSky
January 27, 2025 at 1:11 PM
The contribution of natural transformation for the acquisition of novel genes has been notoriously difficult to quantify because it relies on recombination (which is affected by other processes). Here's a first estimate : doi.org/10.1101/2025... (for the very busy: 1-6% of gene gains) #MicroSky
Reposted by Marco Molari
Building on the UShER tree of millions of SARS-CoV-2 genomes maintained by Angie Hinrichs, Hugh Haddox and Georg Angehrn (and others in @matsen.bsky.social lab and @jbloomlab.bsky.social) have looked into how the neutral mutation rate varies along the genome:
[1/N]
www.biorxiv.org/content/10.1...
[1/N]
www.biorxiv.org/content/10.1...

The mutation rate of SARS-CoV-2 is highly variable between sites and is influenced by sequence context, genomic region, and RNA structure
RNA viruses like SARS-CoV-2 have a high mutation rate, which contributes to their rapid evolution. The rate of mutations depends on the mutation type (e.g., A→C, A→G, etc.) and can vary between sites ...
www.biorxiv.org
January 11, 2025 at 5:37 PM
Building on the UShER tree of millions of SARS-CoV-2 genomes maintained by Angie Hinrichs, Hugh Haddox and Georg Angehrn (and others in @matsen.bsky.social lab and @jbloomlab.bsky.social) have looked into how the neutral mutation rate varies along the genome:
[1/N]
www.biorxiv.org/content/10.1...
[1/N]
www.biorxiv.org/content/10.1...
Thrilled to announce that our work (w. with fantastic @neher.io and Liam Shaw) has been published on Molecular Biology and Evolution! 🎉
academic.oup.com/mbe/advance-...
Are you curious about how fast the genome of E.coli evolves structurally (gains, rearrangements...) ? 🧬
A summary thread [1/N]🧵
academic.oup.com/mbe/advance-...
Are you curious about how fast the genome of E.coli evolves structurally (gains, rearrangements...) ? 🧬
A summary thread [1/N]🧵

Quantifying the evolutionary dynamics of structure and content in closely-related E. coli genomes
Abstract. Bacterial genomes primarily diversify via gain, loss, and rearrangement of genetic material in their flexible accessory genome. Yet the dynamics
academic.oup.com
January 6, 2025 at 5:12 PM
Thrilled to announce that our work (w. with fantastic @neher.io and Liam Shaw) has been published on Molecular Biology and Evolution! 🎉
academic.oup.com/mbe/advance-...
Are you curious about how fast the genome of E.coli evolves structurally (gains, rearrangements...) ? 🧬
A summary thread [1/N]🧵
academic.oup.com/mbe/advance-...
Are you curious about how fast the genome of E.coli evolves structurally (gains, rearrangements...) ? 🧬
A summary thread [1/N]🧵
Reposted by Marco Molari
It's a huge honour to have been named by Nature as "One to Watch" in 2025 for
@pathoplexus.org ! 🙏
I'm proud to see Pathoplexus continuing to draw attention - but this is truly a team effort and only possible thanks to all the amazing people involved! 🙌
swisstph.ch/en/news/news...
1/2
@pathoplexus.org ! 🙏
I'm proud to see Pathoplexus continuing to draw attention - but this is truly a team effort and only possible thanks to all the amazing people involved! 🙌
swisstph.ch/en/news/news...
1/2

Swiss TPH Researcher Emma Hodcroft Named by Nature as 1 of 3 People To Watch in 2025
Emma Hodcroft, group leader at Swiss TPH and assistant professor at the University of Basel, was named one of three people to watch in shaping science in 2025 by the renowned...
swisstph.ch
December 11, 2024 at 11:14 AM
It's a huge honour to have been named by Nature as "One to Watch" in 2025 for
@pathoplexus.org ! 🙏
I'm proud to see Pathoplexus continuing to draw attention - but this is truly a team effort and only possible thanks to all the amazing people involved! 🙌
swisstph.ch/en/news/news...
1/2
@pathoplexus.org ! 🙏
I'm proud to see Pathoplexus continuing to draw attention - but this is truly a team effort and only possible thanks to all the amazing people involved! 🙌
swisstph.ch/en/news/news...
1/2
Reposted by Marco Molari
It's an incredible honour to have received this #OpenData prize from Swiss Academies of Arts & Sciences for @pathoplexus.org!
Though not everyone in Pathoplexus could be there in person, it was a fantastic & thought-provoking event, & I'm so glad were part of it! #OpenScience #OpenSource
Though not everyone in Pathoplexus could be there in person, it was a fantastic & thought-provoking event, & I'm so glad were part of it! #OpenScience #OpenSource

November 28, 2024 at 4:56 PM
It's an incredible honour to have received this #OpenData prize from Swiss Academies of Arts & Sciences for @pathoplexus.org!
Though not everyone in Pathoplexus could be there in person, it was a fantastic & thought-provoking event, & I'm so glad were part of it! #OpenScience #OpenSource
Though not everyone in Pathoplexus could be there in person, it was a fantastic & thought-provoking event, & I'm so glad were part of it! #OpenScience #OpenSource