Arjun Biddanda
@aabiddanda.github.io
370 followers
410 following
10 posts
Statistical and population geneticist | Postdoc @ JHU Biology | COYS | Costco enthusiast | aabiddanda.github.io
Posts
Media
Videos
Starter Packs
Pinned

Arjun Biddanda
@aabiddanda.github.io
· Apr 7
Reposted by Arjun Biddanda







Reposted by Arjun Biddanda


Reposted by Arjun Biddanda

Reposted by Arjun Biddanda

Reposted by Arjun Biddanda
Reposted by Arjun Biddanda


Reposted by Arjun Biddanda

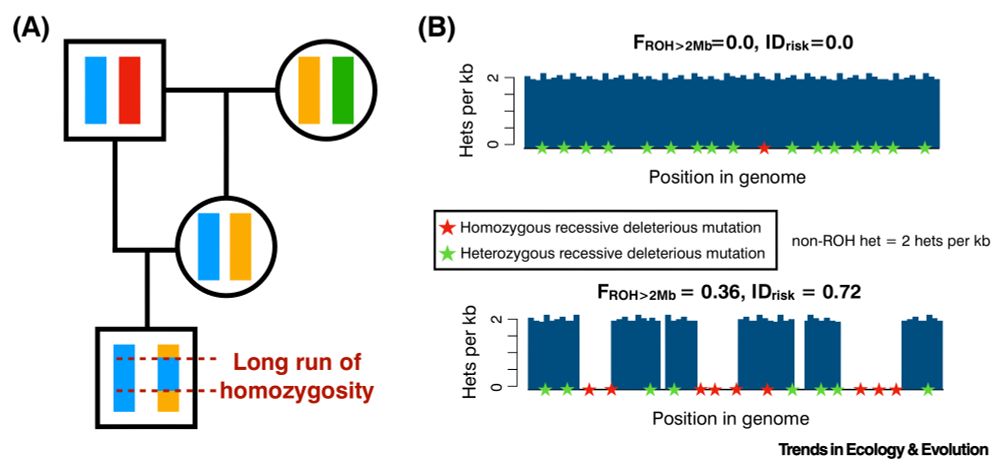
![Comic. [Building with large sign in front of it[ SIGN: Welcome to the *Biology Department* It has been [changeable sign: 3] days since we discovered something existentially horrifying about bugs that makes you question your whole reality](https://cdn.bsky.app/img/feed_thumbnail/plain/did:plc:cz73r7iyiqn26upot4jtjdhk/bafkreigvmzverlrjgqp4mazbkhm43fpvv2hdbkd2t2646dhihozrwdcy24@jpeg)
Reposted by Arjun Biddanda

Yun S. Song
@yun-s-song.bsky.social
· Sep 11

Robust and accurate Bayesian inference of genome-wide genealogies for hundreds of genomes - Nature Genetics
SINGER is a method for creating ancestral recombination graphs to understand the genealogical history of genomes. The method has increased speed, and thus scalability, without sacrificing accuracy.
doi.org
Reposted by Arjun Biddanda


Reposted by Arjun Biddanda


Reposted by Arjun Biddanda

Joao Ascensao
@joaoascensao.bsky.social
· Aug 21

Frequency-dependent fitness effects are ubiquitous
In simple microbial populations, the fitness effects of most selected mutations are generally taken to be constant, independent of genotype frequency. This assumption underpins predictions about evolutionary dynamics, epistatic interactions, and the maintenance of genetic diversity in populations. Here, we systematically test this assumption using beneficial mutations from early generations of the Escherichia coli Long-Term Evolution Experiment (LTEE). Using flow cytometry-based competition assays, we find that frequency-dependent fitness effects are the norm rather than the exception, occurring in approximately 80\% of strain pairs tested. Most competitions exhibit negative frequency-dependence, where fitness advantages decline as mutant frequency increases. Furthermore, we demonstrate that the strength of frequency-dependence is predictable from invasion fitness measurements, with invasion fitness explaining approximately half of the biological variation in frequency-dependent slopes. Additionally, we observe violations of fitness transitivity in several strain combinations, indicating that competitive relationships cannot always be predicted from fitness relative to a single reference strain alone. Through high-resolution measurements of within-growth cycle dynamics, we show that simple resource competition explains a substantial portion of the frequency-dependence: when faster-growing genotypes dominate populations, they deplete shared resources more rapidly, reducing the time available for fitness differences to accumulate. Our results demonstrate that even in a simple model system designed to minimize ecological complexity, subtle ecological interactions between closely related genotypes create frequency-dependent selection that can fundamentally alter evolutionary dynamics. ### Competing Interest Statement The authors have declared no competing interest.
doi.org


Reposted by Arjun Biddanda

Carly
@nautiluscarly.bsky.social
· Aug 19
Reposted by Arjun Biddanda

James Kitchens
@kitchensjn.bsky.social
· Aug 19

tskit_arg_visualizer: interactive plotting of ancestral recombination graphs
Summary: Ancestral recombination graphs (ARGs) are a complete representation of the genetic relationships between recombining lineages and are of central importance in population genetics. Recent brea...
arxiv.org
Reposted by Arjun Biddanda



Reposted by Arjun Biddanda


Reposted by Arjun Biddanda




Reposted by Arjun Biddanda


Reposted by Arjun Biddanda

Reposted by Arjun Biddanda