Alex Kalinin
@alxndrkalinin.bsky.social
250 followers
250 following
25 posts
AI/ML for image-based profiling @broadinstitute.org | prev CUHK-SZ & UMich
Posts
Media
Videos
Starter Packs
Pinned


Reposted by Alex Kalinin

Reposted by Alex Kalinin


Alex Kalinin
@alxndrkalinin.bsky.social
· Jul 12
Alex Kalinin
@alxndrkalinin.bsky.social
· Jul 10

Foreground-aware Virtual Staining for Accurate 3D Cell Morphological Profiling
Microscopy enables direct observation of cellular morphology in 3D, with transmitted-light methods offering low-cost, minimally invasive imaging and fluorescence microscopy providing specificity and c...
arxiv.org

Alex Kalinin
@alxndrkalinin.bsky.social
· Jul 10
Alex Kalinin
@alxndrkalinin.bsky.social
· Jul 10
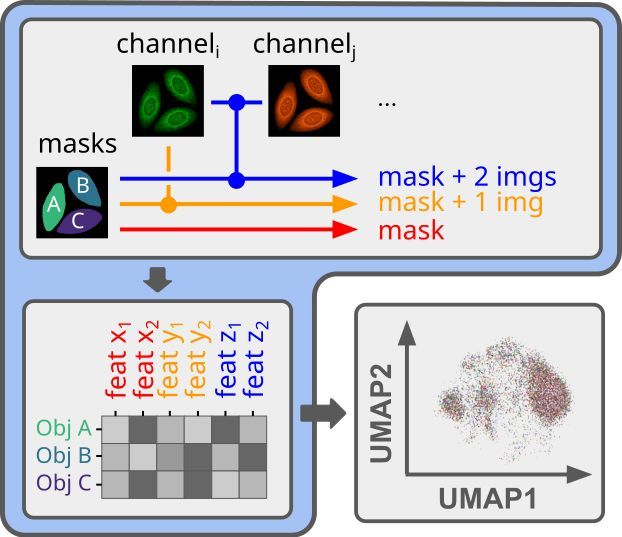
Reposted by Alex Kalinin

Reposted by Alex Kalinin


Alex Kalinin
@alxndrkalinin.bsky.social
· Jun 23


Reposted by Alex Kalinin


Alex Kalinin
@alxndrkalinin.bsky.social
· Jun 10

Alex Kalinin
@alxndrkalinin.bsky.social
· Jun 10
Alex Kalinin
@alxndrkalinin.bsky.social
· Jun 10
