Diego del Alamo
@delalamo.xyz
3.2K followers
2K following
1.8K posts
Computational protein engineering & synthetic biochemistry
Opinions my own
https://linktr.ee/ddelalamo
Posts
Media
Videos
Starter Packs
Pinned

Diego del Alamo
@delalamo.xyz
· May 15

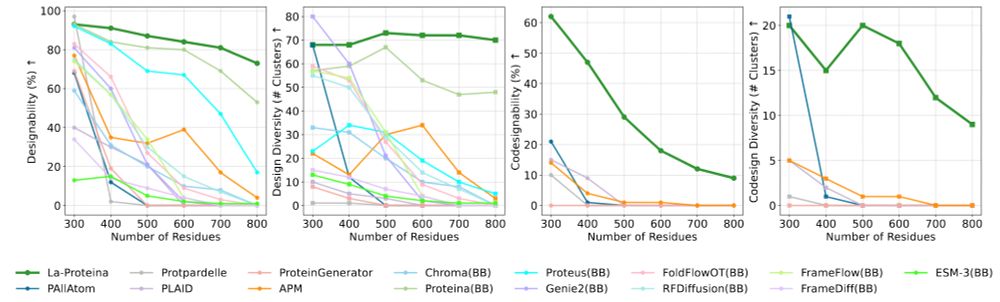
Adapting ProteinMPNN for antibody design without retraining
The neural network ProteinMPNN designs protein sequences capable of folding into predefined tertiary structures and quaternary assemblies. It has become widely used due to its high success rates when ...
www.biorxiv.org








Reposted by Diego del Alamo


Diego del Alamo
@delalamo.xyz
· 12d


Reposted by Diego del Alamo


Diego del Alamo
@delalamo.xyz
· 14d
Diego del Alamo
@delalamo.xyz
· 18d
Diego del Alamo
@delalamo.xyz
· 18d


Diego del Alamo
@delalamo.xyz
· 18d
Reposted by Diego del Alamo




Diego del Alamo
@delalamo.xyz
· 19d
Reposted by Diego del Alamo