Markus List
@itisalist.bsky.social
290 followers
530 following
27 posts
Assistant Professor for Data Science in Systems Biology at the Technical University of Munich (http://daisybio.de). Mostly posting about bioinformatics and systems / network biology research. Views are my own. he / him.
Posts
Media
Videos
Starter Packs

Reposted by Markus List









Reposted by Markus List


Reposted by Markus List



Markus List
@itisalist.bsky.social
· Jun 5





Markus List
@itisalist.bsky.social
· May 27
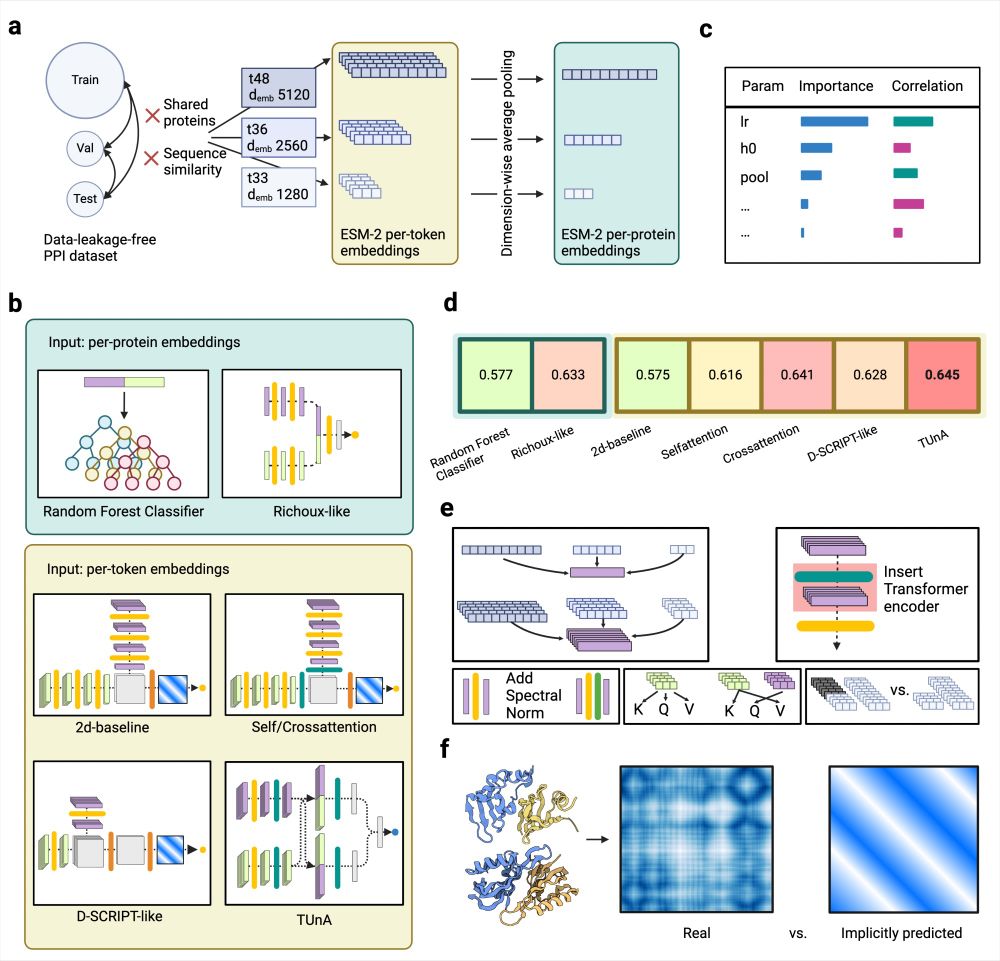
Guiding questions to avoid data leakage in biological machine learning applications - Nature Methods
This Perspective discusses the issue of data leakage in machine learning based models and presents seven questions designed to identify and avoid the problems resulting from data leakage.
www.nature.com

Reposted by Markus List

Victor Javier
@vjsanchez.bsky.social
· Apr 30
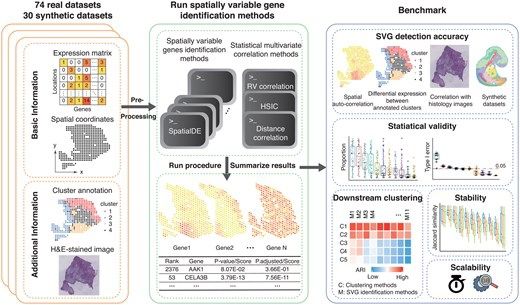
Benchmarking algorithms for spatially variable gene identification in spatial transcriptomics
AbstractMotivation. The rapid development of spatial transcriptomics has underscored the importance of identifying spatially variable genes. As a fundament
academic.oup.com
Reposted by Markus List





Reposted by Markus List

Ana Conesa
@anaconesa.bsky.social
· Mar 28

Reposted by Markus List

Markus List
@itisalist.bsky.social
· Mar 3
Reposted by Markus List


Reposted by Markus List
DaiSyBio
@daisybio.de
· Jan 30

Johannes Kersting Awarded an ASAPBio Poster Prize for RExPO Contribution
The ASAPBio poster competition is an initiative that champions open science by highlighting innovative research and the early sharing of results. This year’s competition emphasized transparency, colla...
blog.scienceopen.com