GAMA Miguel Angel
@miangoar.bsky.social
220 followers
190 following
93 posts
Biologist that navigate in the oceans of diversity through space-time
Protein evolution, metagenomics, AI/ML/DL
Website https://miangoaren.github.io/
Posts
Media
Videos
Starter Packs


Reposted by GAMA Miguel Angel



Reposted by GAMA Miguel Angel

Sam Berry
@sberry.bsky.social
· Aug 26
Reposted by GAMA Miguel Angel







Reposted by GAMA Miguel Angel


GAMA Miguel Angel
@miangoar.bsky.social
· Aug 27
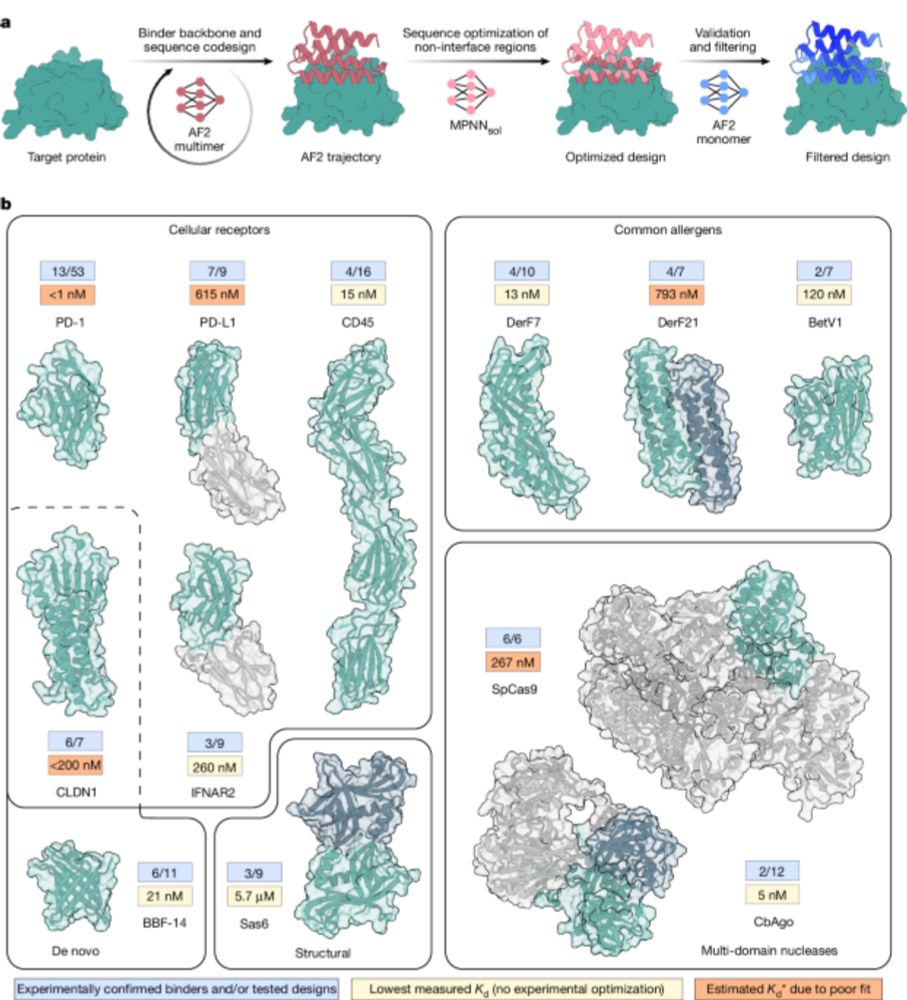
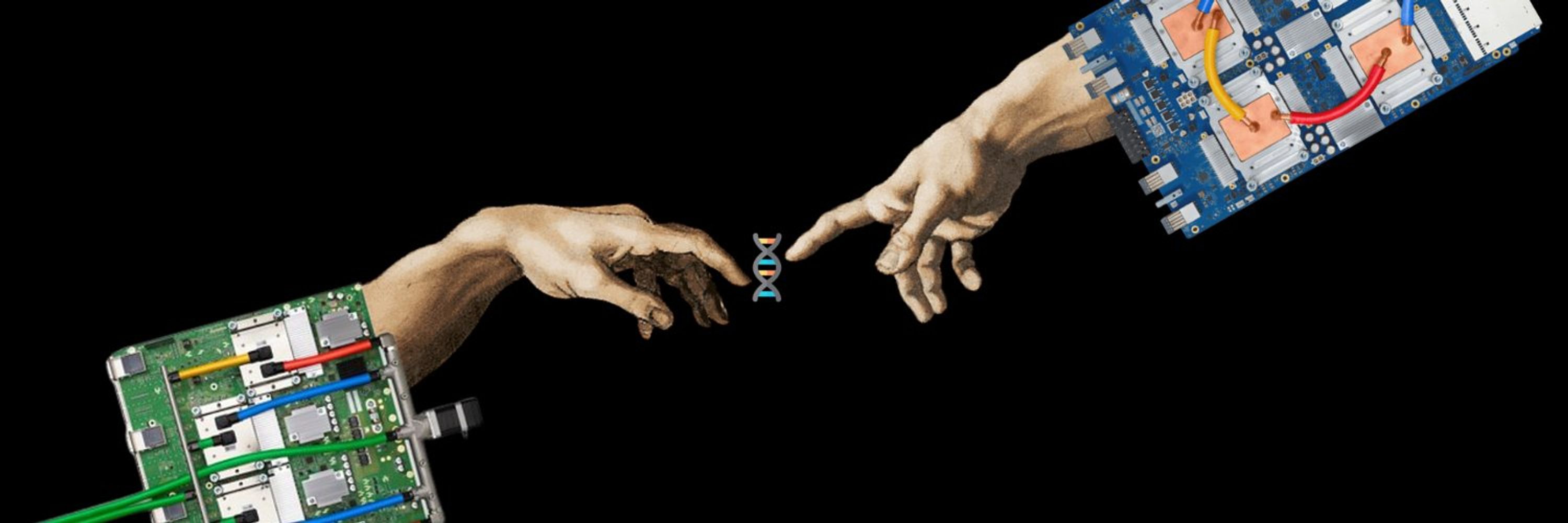
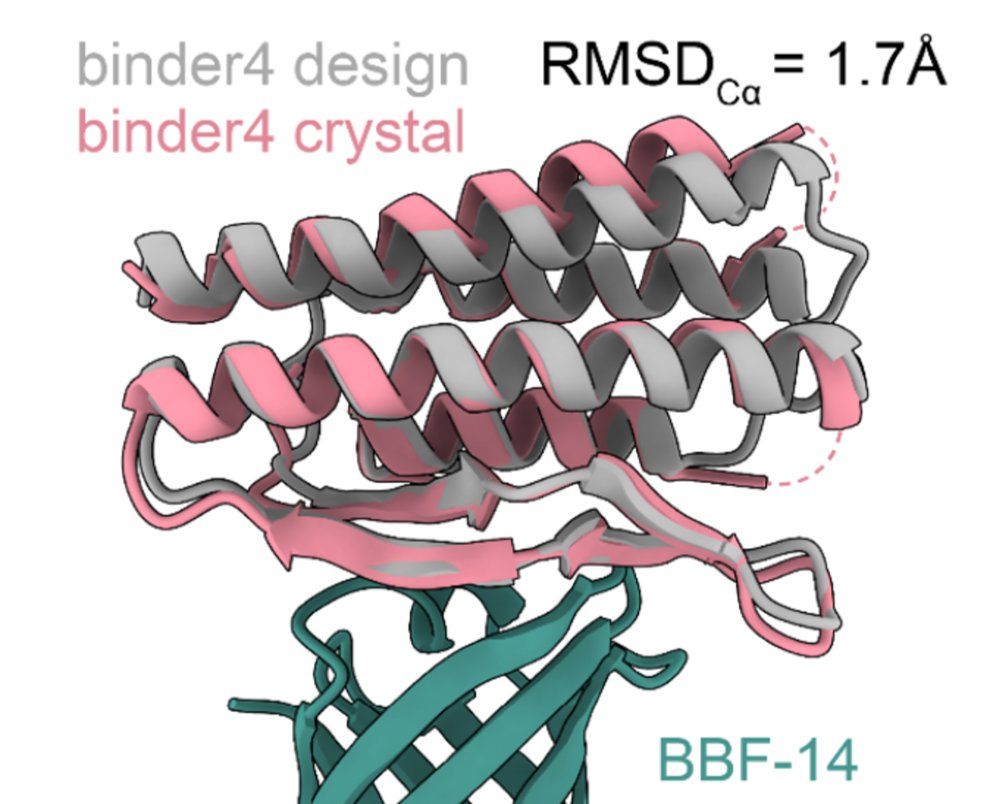
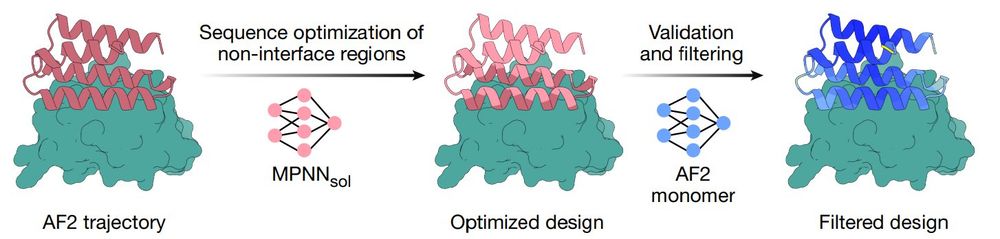
One-shot design of functional protein binders with BindCraft - Nature
BindCraft, an open-source, automated pipeline for de novo protein binder design with experimental success rates of 10–100%, leverages AlphaFold2 weights to generate binders with nanomolar affinity wit...
nature.com

GAMA Miguel Angel
@miangoar.bsky.social
· Aug 27

Adaptyv Bio - Protein Design Competition: Has binder design been solved?
We analyze the results of our protein design competition where 130 designers created binders for EGFR. With a 5x improvement in success rates and some designs outperforming clinical antibodies, we exp...
adaptyvbio.com







GAMA Miguel Angel
@miangoar.bsky.social
· Aug 27
GAMA Miguel Angel
@miangoar.bsky.social
· Aug 27

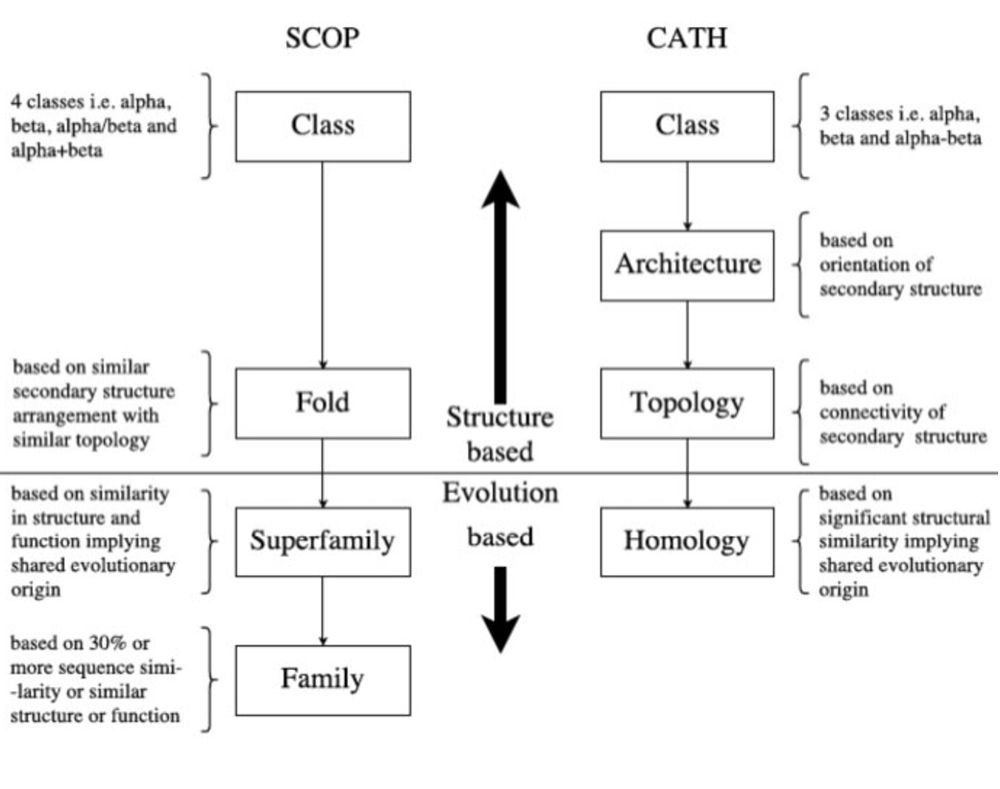
ECOD: integrating classifications of protein domains from experimental and predicted structures - PubMed
The evolutionary classification of protein domains (ECOD) classifies protein domains using a combination of sequence and structural data (http://prodata.swmed.edu/ecod). Here we present the culminatio...
pubmed.ncbi.nlm.nih.gov
Reposted by GAMA Miguel Angel

