




Gaëtan Benoit
@gaetanbenoit.bsky.social
Postdoc researcher in bioinformatics at Pasteur institute. Scalable methods and software for metagenomics. https://github.com/GaetanBenoitDev
Reposted by Gaëtan Benoit

Ok; mim (github.com/COMBINE-lab/...) preprint submitted! Excited for folks to see it and share thoughts. The key takeaway; mim allows the quick, one-time, building of a small auxiliary index that then allows scaling gzipped FASTQ parsing linearly in # of threads. 1/2

GitHub - COMBINE-lab/mim: A small, auxiliary index to massively improve parallel fastq parsing
A small, auxiliary index to massively improve parallel fastq parsing - COMBINE-lab/mim
github.com
November 25, 2025 at 2:13 PM
Ok; mim (github.com/COMBINE-lab/...) preprint submitted! Excited for folks to see it and share thoughts. The key takeaway; mim allows the quick, one-time, building of a small auxiliary index that then allows scaling gzipped FASTQ parsing linearly in # of threads. 1/2
Reposted by Gaëtan Benoit

Yohan Hernandez–Courbevoie presenting REINDEER2 at Seqbim!
For those who missed it, the introduction thread of REINDEER2
bsky.app/profile/npma...
For those who missed it, the introduction thread of REINDEER2
bsky.app/profile/npma...

November 24, 2025 at 12:41 PM
Yohan Hernandez–Courbevoie presenting REINDEER2 at Seqbim!
For those who missed it, the introduction thread of REINDEER2
bsky.app/profile/npma...
For those who missed it, the introduction thread of REINDEER2
bsky.app/profile/npma...
Reposted by Gaëtan Benoit
@wytamma.bsky.social : so, it took a little bit of extra time (not the flight back from the CZI meeting), but I decided to just f#&$ing do it, and the basic code to build and parse with the auxiliary fastq index is working (github.com/COMBINE-lab/...). 1/2

GitHub - COMBINE-lab/mim: A small, auxiliary index to massively improve parallel fastq parsing
A small, auxiliary index to massively improve parallel fastq parsing - COMBINE-lab/mim
github.com
November 19, 2025 at 3:01 AM
@wytamma.bsky.social : so, it took a little bit of extra time (not the flight back from the CZI meeting), but I decided to just f#&$ing do it, and the basic code to build and parse with the auxiliary fastq index is working (github.com/COMBINE-lab/...). 1/2
Reposted by Gaëtan Benoit

“Bin Chicken” is now published in Nature Methods! It substantially improves genome recovery through rational coassembly 🧬🖥️. Applied to public 🌍 metagenomes, we recovered 24,000 novel species 🦠, including 6 new phyla.
doi.org/10.1038/s415...
@benjwoodcroft.bsky.social @rhysnewell.bsky.social
🧵1/6
doi.org/10.1038/s415...
@benjwoodcroft.bsky.social @rhysnewell.bsky.social
🧵1/6

November 13, 2025 at 10:09 AM
“Bin Chicken” is now published in Nature Methods! It substantially improves genome recovery through rational coassembly 🧬🖥️. Applied to public 🌍 metagenomes, we recovered 24,000 novel species 🦠, including 6 new phyla.
doi.org/10.1038/s415...
@benjwoodcroft.bsky.social @rhysnewell.bsky.social
🧵1/6
doi.org/10.1038/s415...
@benjwoodcroft.bsky.social @rhysnewell.bsky.social
🧵1/6
Reposted by Gaëtan Benoit

Metagenomics colleagues!
I'm looking for studies where both Illumina and ONT sequencing were performed on the same samples from soil, human, ruminent, and other sample types for comparison. Bonus if those studies include PacBio data.
Please help and share!
I'm looking for studies where both Illumina and ONT sequencing were performed on the same samples from soil, human, ruminent, and other sample types for comparison. Bonus if those studies include PacBio data.
Please help and share!
November 11, 2025 at 8:21 PM
Metagenomics colleagues!
I'm looking for studies where both Illumina and ONT sequencing were performed on the same samples from soil, human, ruminent, and other sample types for comparison. Bonus if those studies include PacBio data.
Please help and share!
I'm looking for studies where both Illumina and ONT sequencing were performed on the same samples from soil, human, ruminent, and other sample types for comparison. Bonus if those studies include PacBio data.
Please help and share!
Reposted by Gaëtan Benoit

Our method for genome size estimation from long-read overlaps is now published 🥳
academic.oup.com/bioinformati...
academic.oup.com/bioinformati...

Genome size estimation from long read overlaps
AbstractMotivation. Accurate genome size estimation is an important component of genomic analyses such as assembly and coverage calculation, though existin
academic.oup.com
November 7, 2025 at 3:19 AM
Our method for genome size estimation from long-read overlaps is now published 🥳
academic.oup.com/bioinformati...
academic.oup.com/bioinformati...
Reposted by Gaëtan Benoit

1/6 Movi 2 is here: faster and more space-efficient for pangenome queries. Its fastest mode uses half the memory of Movi 1 while running ~30% faster. github.com/mohsenzakeri...

GitHub - mohsenzakeri/Movi: Fast, Cache-Efficient, and Scalable Queries on Pangenomes
Fast, Cache-Efficient, and Scalable Queries on Pangenomes - mohsenzakeri/Movi
github.com
October 21, 2025 at 8:00 PM
1/6 Movi 2 is here: faster and more space-efficient for pangenome queries. Its fastest mode uses half the memory of Movi 1 while running ~30% faster. github.com/mohsenzakeri...
Reposted by Gaëtan Benoit

Ca n'est pas si souvent, un article publié dans Nature met ma communauté à l'honneur (la bioinformatique des séquences). Je vous raconte ?
www.nature.com/articles/d41...
www.nature.com/articles/d41...

‘Google for DNA’ brings order to biology’s big data
MetaGraph compresses vast data archives into a search engine for scientists, opening up new frontiers of biological discovery.
www.nature.com
October 9, 2025 at 3:00 PM
Ca n'est pas si souvent, un article publié dans Nature met ma communauté à l'honneur (la bioinformatique des séquences). Je vous raconte ?
www.nature.com/articles/d41...
www.nature.com/articles/d41...
Reposted by Gaëtan Benoit

Our preprint on our new metagenomic HiFi assembler Alice is out 🥳 Based on a *new sketching method* (🧵1/6)
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...

Alice: fast and haplotype-aware assembly of high-fidelity reads based on MSR sketching
We introduce Mapping-friendly Sequence Reduction (MSR) sketches, a sketching method for high-fidelity (HiFi) long reads, and Alice, an assembler that operates directly on these sketches. MSR produces ...
www.biorxiv.org
October 3, 2025 at 2:51 PM
Our preprint on our new metagenomic HiFi assembler Alice is out 🥳 Based on a *new sketching method* (🧵1/6)
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
👉 Preprint www.biorxiv.org/content/10.1...
👉 Github github.com/rolandfaure/...
Reposted by Gaëtan Benoit

New pre-print from the Banfield lab, highlighting an interesting case of 1.5Mb megaplasmids found in human gut.
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...

Megaplasmids associate with Escherichia coli and other Enterobacteriaceae
Humans and animals are ubiquitously colonized by Enterobacteriaceae , a bacterial family that contains both commensals and clinically significant pathogens. Here, we report Enterobacteriaceae megaplas...
doi.org
October 1, 2025 at 4:44 PM
New pre-print from the Banfield lab, highlighting an interesting case of 1.5Mb megaplasmids found in human gut.
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...
Plasmid genomes were resolved using #PacBio HiFi sequencing with hifiasm-meta for #metagenome assembly. Host association was detected using epigenetic signals.
doi.org/10.1101/2025...
Reposted by Gaëtan Benoit

Alice: fast and haplotype-aware assembly of high-fidelity reads based on MSR sketching https://www.biorxiv.org/content/10.1101/2025.09.29.679204v1
October 1, 2025 at 1:47 AM
Alice: fast and haplotype-aware assembly of high-fidelity reads based on MSR sketching https://www.biorxiv.org/content/10.1101/2025.09.29.679204v1

Reposted by Gaëtan Benoit

Delighted to see our paper studying the evolution of plasmids over the last 100 years, now out! Years of work by Adrian Cazares, also Nick Thomson @sangerinstitute.bsky.social - this version much improved over the preprint. Final version should be open access, apols.
Thread 1/n
Thread 1/n

September 25, 2025 at 9:29 PM
Delighted to see our paper studying the evolution of plasmids over the last 100 years, now out! Years of work by Adrian Cazares, also Nick Thomson @sangerinstitute.bsky.social - this version much improved over the preprint. Final version should be open access, apols.
Thread 1/n
Thread 1/n
Reposted by Gaëtan Benoit

Delighted to finally announce a preprint describing the Q100 project! “A complete diploid human genome benchmark for personalized genomics” For which we finished HG002 to near-perfect accuracy: www.biorxiv.org/content/10.1... 🧵[1/14]

A complete diploid human genome benchmark for personalized genomics
Human genome resequencing typically involves mapping reads to a reference genome to call variants; however, this approach suffers from both technical and reference biases, leaving many duplicated and ...
www.biorxiv.org
September 22, 2025 at 5:01 PM
Delighted to finally announce a preprint describing the Q100 project! “A complete diploid human genome benchmark for personalized genomics” For which we finished HG002 to near-perfect accuracy: www.biorxiv.org/content/10.1... 🧵[1/14]
Reposted by Gaëtan Benoit

Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
High-resolution metagenome assembly for modern long reads with myloasm https://www.biorxiv.org/content/10.1101/2025.09.05.674543v1
September 7, 2025 at 11:35 PM
Preprint out for myloasm, our new nanopore / HiFi metagenome assembler!
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Nanopore's getting accurate, but
1. Can this lead to better metagenome assemblies?
2. How, algorithmically, to leverage them?
with co-author Max Marin @mgmarin.bsky.social, supervised by Heng Li @lh3lh3.bsky.social
1 / N
Reposted by Gaëtan Benoit

New blog post!
metaMDBG (@gaetanbenoit.bsky.social) and Myloasm (@jimshaw.bsky.social) have had recent releases, so I updated the benchmarks from the Autocycler paper:
rrwick.github.io/2025/09/23/a...
Both tools improved considerably! Time to update your conda environments 😄
metaMDBG (@gaetanbenoit.bsky.social) and Myloasm (@jimshaw.bsky.social) have had recent releases, so I updated the benchmarks from the Autocycler paper:
rrwick.github.io/2025/09/23/a...
Both tools improved considerably! Time to update your conda environments 😄
Benchmark update: metaMDBG and Myloasm
a blog for miscellaneous bioinformatics stuff
rrwick.github.io
September 23, 2025 at 1:53 AM
New blog post!
metaMDBG (@gaetanbenoit.bsky.social) and Myloasm (@jimshaw.bsky.social) have had recent releases, so I updated the benchmarks from the Autocycler paper:
rrwick.github.io/2025/09/23/a...
Both tools improved considerably! Time to update your conda environments 😄
metaMDBG (@gaetanbenoit.bsky.social) and Myloasm (@jimshaw.bsky.social) have had recent releases, so I updated the benchmarks from the Autocycler paper:
rrwick.github.io/2025/09/23/a...
Both tools improved considerably! Time to update your conda environments 😄
Reposted by Gaëtan Benoit
Achievement unlocked: defend your habilitation thesis on the same day than your partner. That was quite a science + celebration day, thanks to all involved 💙✨


September 5, 2025 at 4:51 PM
Achievement unlocked: defend your habilitation thesis on the same day than your partner. That was quite a science + celebration day, thanks to all involved 💙✨
Reposted by Gaëtan Benoit

🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...

September 3, 2025 at 8:39 AM
🌎👩🔬 For 15+ years biology has accumulated petabytes (million gigabytes) of🧬DNA sequencing data🧬 from the far reaches of our planet.🦠🍄🌵
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Logan now democratizes efficient access to the world’s most comprehensive genetics dataset. Free and open.
doi.org/10.1101/2024...
Reposted by Gaëtan Benoit
skani v0.3.0 is released. github.com/bluenote-157...
- 30-40% potential reduction in memory with approximately the same runtime.
- Breaking changes to indexing and searching databases
Calculate ANI for contigs, genomes -- even search > 140k genomes. Pre-indexed GTDB-R226 available for download.
- 30-40% potential reduction in memory with approximately the same runtime.
- Breaking changes to indexing and searching databases
Calculate ANI for contigs, genomes -- even search > 140k genomes. Pre-indexed GTDB-R226 available for download.

GitHub - bluenote-1577/skani: Fast, robust ANI and aligned fraction for (metagenomic) genomes and contigs.
Fast, robust ANI and aligned fraction for (metagenomic) genomes and contigs. - bluenote-1577/skani
github.com
August 13, 2025 at 2:19 PM
skani v0.3.0 is released. github.com/bluenote-157...
- 30-40% potential reduction in memory with approximately the same runtime.
- Breaking changes to indexing and searching databases
Calculate ANI for contigs, genomes -- even search > 140k genomes. Pre-indexed GTDB-R226 available for download.
- 30-40% potential reduction in memory with approximately the same runtime.
- Breaking changes to indexing and searching databases
Calculate ANI for contigs, genomes -- even search > 140k genomes. Pre-indexed GTDB-R226 available for download.
Reposted by Gaëtan Benoit
Longdust, a new tool to identify highly repetitive STRs, VNTRs, satellite DNA and other low-complexity regions (LCRs). Similar to SDUST but for long regions.
github.com/lh3/longdust
github.com/lh3/longdust
GitHub - lh3/longdust: Identify long STRs, VNTRs, satellite DNA and other low-complexity regions in a genome
Identify long STRs, VNTRs, satellite DNA and other low-complexity regions in a genome - lh3/longdust
github.com
July 31, 2025 at 7:59 PM
Longdust, a new tool to identify highly repetitive STRs, VNTRs, satellite DNA and other low-complexity regions (LCRs). Similar to SDUST but for long regions.
github.com/lh3/longdust
github.com/lh3/longdust
Reposted by Gaëtan Benoit

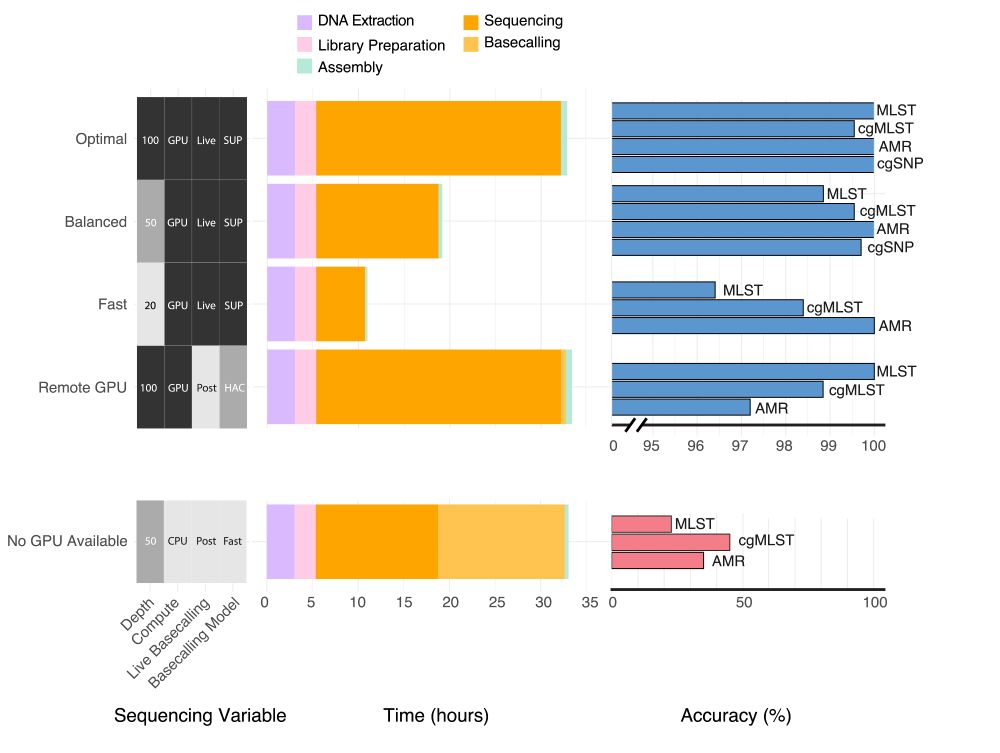
Pleased to say that our preprint benchmarking Nanopore data for MLST, cgMLST, cgSNP & AMR typing from bacterial isolates is out! TL;DR you can get almost perfect results from 50x depth using live SUP basecalling with a GPU in under 20 hours #microsky#IDsky 🦠🧬🖥️ /1
www.medrxiv.org/content/10.1...
www.medrxiv.org/content/10.1...
July 30, 2025 at 2:11 AM
Pleased to say that our preprint benchmarking Nanopore data for MLST, cgMLST, cgSNP & AMR typing from bacterial isolates is out! TL;DR you can get almost perfect results from 50x depth using live SUP basecalling with a GPU in under 20 hours #microsky#IDsky 🦠🧬🖥️ /1
www.medrxiv.org/content/10.1...
www.medrxiv.org/content/10.1...
Reposted by Gaëtan Benoit

Congratulations to Rayan Chiki, (Institut Pasteur) head of the “Sequence Bioinformatics” unit, for securing the ERC Proof of Concept 2025 for his project ENZYMINER! 👏
@rayan.chiki.bsky.social
#Bioinformatics
@rayan.chiki.bsky.social
#Bioinformatics


July 24, 2025 at 3:10 PM
Congratulations to Rayan Chiki, (Institut Pasteur) head of the “Sequence Bioinformatics” unit, for securing the ERC Proof of Concept 2025 for his project ENZYMINER! 👏
@rayan.chiki.bsky.social
#Bioinformatics
@rayan.chiki.bsky.social
#Bioinformatics
Reposted by Gaëtan Benoit
New sylph pre-built databases + taxonomy available for:
- GTDB-R226 (143k prok. species)
- GlobDB-R226 (>300k prok. species, thanks @daanspeth.bsky.social )
- UHGV (Unified Human Gut Virome Catalog, thanks @apcamargo.bsky.social )
Must update sylph-tax; see docs (sylph-docs.github.io/sylph-tax/)
- GTDB-R226 (143k prok. species)
- GlobDB-R226 (>300k prok. species, thanks @daanspeth.bsky.social )
- UHGV (Unified Human Gut Virome Catalog, thanks @apcamargo.bsky.social )
Must update sylph-tax; see docs (sylph-docs.github.io/sylph-tax/)
Sylph-tax - Documentation for sylph - ultrafast, precise metagenomic profiling
sylph-docs.github.io
July 23, 2025 at 2:11 PM
New sylph pre-built databases + taxonomy available for:
- GTDB-R226 (143k prok. species)
- GlobDB-R226 (>300k prok. species, thanks @daanspeth.bsky.social )
- UHGV (Unified Human Gut Virome Catalog, thanks @apcamargo.bsky.social )
Must update sylph-tax; see docs (sylph-docs.github.io/sylph-tax/)
- GTDB-R226 (143k prok. species)
- GlobDB-R226 (>300k prok. species, thanks @daanspeth.bsky.social )
- UHGV (Unified Human Gut Virome Catalog, thanks @apcamargo.bsky.social )
Must update sylph-tax; see docs (sylph-docs.github.io/sylph-tax/)
Reposted by Gaëtan Benoit

Two papers in today's issue of @nature.com : 1) we assemble 65 genomes to near completion, including centromeres and the MHC. tinyurl.com/3huhax6w. 2) we sequence 1,019 genomes from the 1kGP with long reads, revealing SVs down to low allele frequencies tinyurl.com/wbx3we9x.
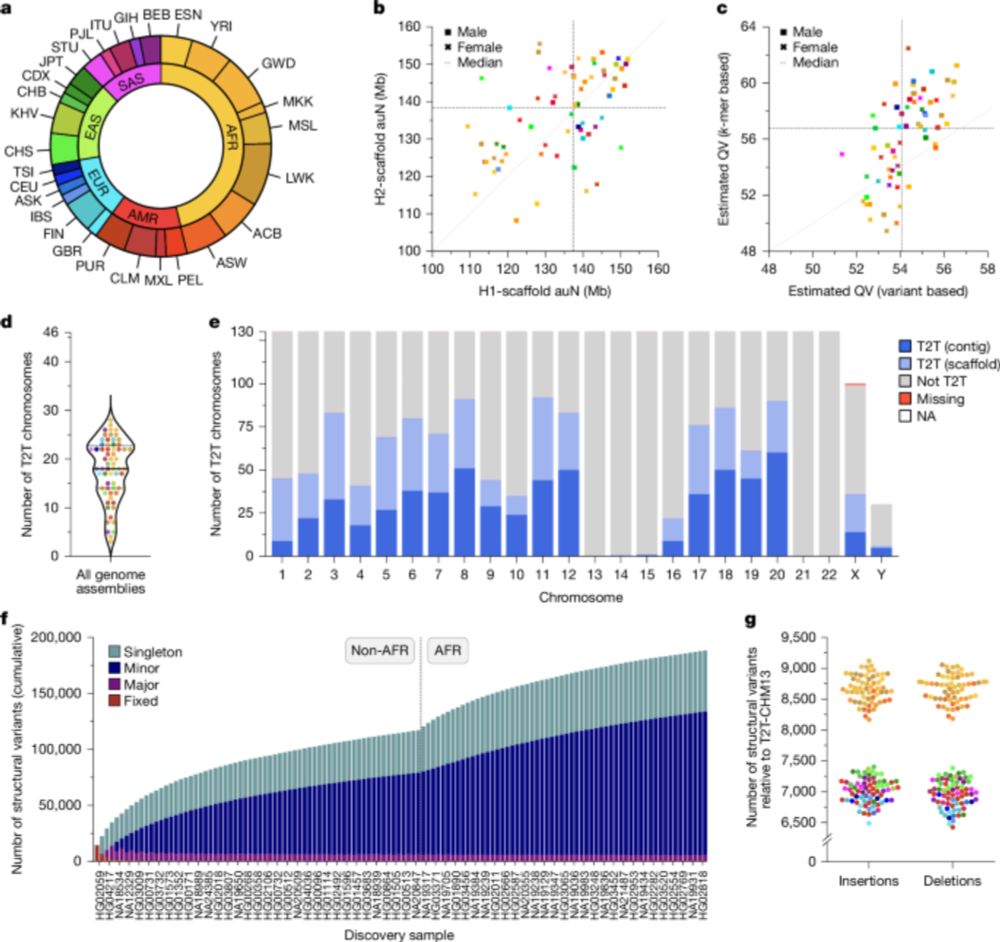
Complex genetic variation in nearly complete human genomes - Nature
Using sequencing and haplotype-resolved assembly of 65 diverse human genomes, complex regions including the major histocompatibility complex and centromeres are analysed.
tinyurl.com
July 23, 2025 at 3:12 PM
Two papers in today's issue of @nature.com : 1) we assemble 65 genomes to near completion, including centromeres and the MHC. tinyurl.com/3huhax6w. 2) we sequence 1,019 genomes from the 1kGP with long reads, revealing SVs down to low allele frequencies tinyurl.com/wbx3we9x.
Reposted by Gaëtan Benoit

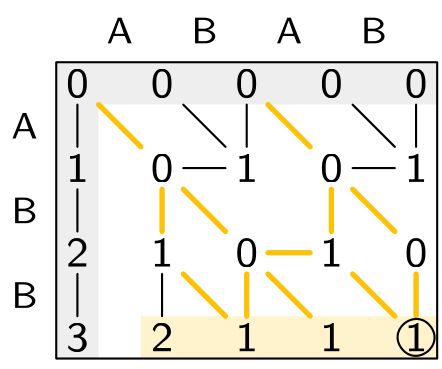
Sassy is out now!
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...
July 18, 2025 at 8:20 PM
Sassy is out now!
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...
Ever need to search for approximate matches of short DNA strings?
Sassy is the tool to use!
Available now wherever you get your code
With @rickbitloo.bsky.social
curiouscoding.nl/papers/sassy...
github.com/ragnarGrootK...