



Jim Shaw
@jimshaw.bsky.social
Postdoc at Dana-Farber and Harvard Med with Heng Li (@lh3lh3.bsky.social). Prev: UBC / UofT.
I like thinking about biological sequence analysis and its applications to metagenomics / microbial genomics.
https://jim-shaw-bluenote.github.io
I like thinking about biological sequence analysis and its applications to metagenomics / microbial genomics.
https://jim-shaw-bluenote.github.io
Reposted by Jim Shaw

Super happy that the AllTheBacteria hypothetical proteins are now in AFDB - hopefully we can start to understand the function of some of them at least 😁

The AlphaFold Database, jointly developed with Google DeepMind, now contains datasets from specialist communities.
This includes rich datasets for microbes, viruses & parasites associated with tropical diseases.
Explore the new datasets in the AlphaFold Database.
www.ebi.ac.uk/about/news/u...
This includes rich datasets for microbes, viruses & parasites associated with tropical diseases.
Explore the new datasets in the AlphaFold Database.
www.ebi.ac.uk/about/news/u...

AlphaFold Database welcomes community datasets
Latest AlphaFold Database update adds high-value datasets for microbial and viral proteins, generated by specialist communities
www.ebi.ac.uk
February 18, 2026 at 4:46 AM
Super happy that the AllTheBacteria hypothetical proteins are now in AFDB - hopefully we can start to understand the function of some of them at least 😁
Reposted by Jim Shaw

I enjoyed reading your post @jimshaw.bsky.social and learning about Savont, thank you for the comparison to Emu and kudos on your new tool!
relevant to points raised in the post, the Emu development team has compiled our follow-up thoughts here: github.com/treangenlab/...
relevant to points raised in the post, the Emu development team has compiled our follow-up thoughts here: github.com/treangenlab/...

Recent updates
Contribute to treangenlab/emu development by creating an account on GitHub.
github.com
February 13, 2026 at 8:02 PM
I enjoyed reading your post @jimshaw.bsky.social and learning about Savont, thank you for the comparison to Emu and kudos on your new tool!
relevant to points raised in the post, the Emu development team has compiled our follow-up thoughts here: github.com/treangenlab/...
relevant to points raised in the post, the Emu development team has compiled our follow-up thoughts here: github.com/treangenlab/...
Reposted by Jim Shaw

A long time ago in a galaxy far away, there was a SARS-CoV-2 pandemic. Our paper, led by @martibartfast.bsky.social
a) correcting errors in 4.5 million genomes & their phylogeny
b) improving representation of the Global South in public data
www.nature.com/articles/s41...
(thread 1/n)
a) correcting errors in 4.5 million genomes & their phylogeny
b) improving representation of the Global South in public data
www.nature.com/articles/s41...
(thread 1/n)

Addressing pandemic-wide systematic errors in the SARS-CoV-2 phylogeny - Nature Methods
This Resource paper presents a global SARS-CoV-2 phylogenetic tree of 4,471,579 high-quality genomes consistently constructed by Viridian, an efficient amplicon-aware assembler.
www.nature.com
February 9, 2026 at 3:16 PM
A long time ago in a galaxy far away, there was a SARS-CoV-2 pandemic. Our paper, led by @martibartfast.bsky.social
a) correcting errors in 4.5 million genomes & their phylogeny
b) improving representation of the Global South in public data
www.nature.com/articles/s41...
(thread 1/n)
a) correcting errors in 4.5 million genomes & their phylogeny
b) improving representation of the Global South in public data
www.nature.com/articles/s41...
(thread 1/n)
Reposted by Jim Shaw

If you have an interest in mixing computation and experiments to understand microbial evolution (for example antibiotic resistance) and you think you might be a good fit for a postdoc in my lab, reach out. If it seems it might be a fit I’m happy to help you frame it as AI for Biology per this call

Holy shit: it's an RFP for the NSF Postdoctoral Research Fellowship in Biology (PRFB). Hello old friend www.nsf.gov/funding/oppo...

Postdoctoral Research Fellowships in Biology (PRFB)
www.nsf.gov
February 6, 2026 at 1:04 AM
If you have an interest in mixing computation and experiments to understand microbial evolution (for example antibiotic resistance) and you think you might be a good fit for a postdoc in my lab, reach out. If it seems it might be a fit I’m happy to help you frame it as AI for Biology per this call
Reposted by Jim Shaw

Now also on arxiv:
arxiv.org/abs/2602.04103
arxiv.org/abs/2602.04103

QuadRank: Engineering a High Throughput Rank
Given a text, a query $\mathsf{rank}(q, c)$ counts the number of occurrences of character $c$ among the first $q$ characters of the text. Space-efficient methods to answer these rank queries form an i...
arxiv.org
February 5, 2026 at 10:53 AM
Now also on arxiv:
arxiv.org/abs/2602.04103
arxiv.org/abs/2602.04103
Reposted by Jim Shaw
The online ANI calculator (skani) that allows you to compare your genome data with reference sequences in GTDB saves so much time and faffing around. :)
GTDB - skani calculator
An interface to compute pairwise ANI of NCBI genomes using the GTDB taxonomy.
gtdb.ecogenomic.org
February 2, 2026 at 8:27 PM
The online ANI calculator (skani) that allows you to compare your genome data with reference sequences in GTDB saves so much time and faffing around. :)
Reposted by Jim Shaw

Thrilled to share our labor of love over the last 5 years 🤩
Leveraging long-read metagenomics (@nanoporetech.com) we identified some of the most prevalent gut phage families that have previously been overlooked in short-read based studies. [1/5]
Read more here: www.biorxiv.org/content/10.6...
Leveraging long-read metagenomics (@nanoporetech.com) we identified some of the most prevalent gut phage families that have previously been overlooked in short-read based studies. [1/5]
Read more here: www.biorxiv.org/content/10.6...
GuFi phages represent the most prevalent viral family-level clusters in the human gut microbiome
Despite being important ecological modulators of the gut microbiome, bacteriophage diversity and function remain under-characterized. We show that short-read metagenomic surveys can miss even globally highly prevalent viral family-level clusters (VFCs), that can be readily assembled and characterized with long-read metagenomic data from a relatively small cohort (n=109). While gut Bacteroidota phages have been the prevailing focus in the literature, we show that highly prevalent gut phage families frequently have Firmicutes hosts (termed GuFi phages), with broad host ranges verified using proximity-ligation (Hi-C) sequencing data. High-throughput sequencing of virus-like particles from fecal samples detected frequent enrichment of GuFi phages across samples, revealing their under-appreciated impact on the gut microbiome. We report the first in vitro induction and imaging of members of prevalent GuFi clades including the candidate orders Heliusvirales , Astravirales (VFC 2) and Suryavirales (VFC 4). Our findings underscore the importance of GuFi phages with broad host ranges in the gut microbiome, and the utility of long-read sequencing for viral discovery, paving the way for deeper insights into the role of bacteriophages in human health and disease. ### Competing Interest Statement IL is an employee of Phase Genomics. National Medical Research Council, 23-0614 National Research Foundation, NRFI09-0015 A*STAR, C210812044
www.biorxiv.org
January 30, 2026 at 10:15 AM
Thrilled to share our labor of love over the last 5 years 🤩
Leveraging long-read metagenomics (@nanoporetech.com) we identified some of the most prevalent gut phage families that have previously been overlooked in short-read based studies. [1/5]
Read more here: www.biorxiv.org/content/10.6...
Leveraging long-read metagenomics (@nanoporetech.com) we identified some of the most prevalent gut phage families that have previously been overlooked in short-read based studies. [1/5]
Read more here: www.biorxiv.org/content/10.6...
Reposted by Jim Shaw

Multiple protein structure alignment at scale with FoldMason | Science https://www.science.org/doi/full/10.1126/science.ads6733?af=R

Multiple protein structure alignment at scale with FoldMason
Protein structure is conserved beyond sequence, making multiple structural alignment (MSTA) essential for analyzing distantly related proteins. Computational prediction methods have vastly extended our repository of available protein structures, ...
www.science.org
January 29, 2026 at 11:31 PM
Multiple protein structure alignment at scale with FoldMason | Science https://www.science.org/doi/full/10.1126/science.ads6733?af=R
Reposted by Jim Shaw

I've just released #rust-htslib 1.0. After a long time with a pretty stable API usage of rust-htslib in production, it feels like the right time to finally move to 1.0. Most important change is probably a switch to thread-safe pointers in BAM record handling. github.com/rust-bio/rus...
January 29, 2026 at 10:37 AM
I've just released #rust-htslib 1.0. After a long time with a pretty stable API usage of rust-htslib in production, it feels like the right time to finally move to 1.0. Most important change is probably a switch to thread-safe pointers in BAM record handling. github.com/rust-bio/rus...
Announcing a new tool for "denoising" long-read amplicon sequences: savont.
Savont enables amplicon sequence variants (ASVs) directly from nanopore (or HiFi) long reads. Tested on 16S nanopore amplicons -- seems to work okay.
1/4
github.com/bluenote-157...
Savont enables amplicon sequence variants (ASVs) directly from nanopore (or HiFi) long reads. Tested on 16S nanopore amplicons -- seems to work okay.
1/4
github.com/bluenote-157...

GitHub - bluenote-1577/savont: Amplicon sequencing variants from 16s ONT R10.4 / HiFi long reads
Amplicon sequencing variants from 16s ONT R10.4 / HiFi long reads - bluenote-1577/savont
github.com
January 28, 2026 at 6:46 PM
Announcing a new tool for "denoising" long-read amplicon sequences: savont.
Savont enables amplicon sequence variants (ASVs) directly from nanopore (or HiFi) long reads. Tested on 16S nanopore amplicons -- seems to work okay.
1/4
github.com/bluenote-157...
Savont enables amplicon sequence variants (ASVs) directly from nanopore (or HiFi) long reads. Tested on 16S nanopore amplicons -- seems to work okay.
1/4
github.com/bluenote-157...
Reposted by Jim Shaw

New blog post with some thoughts on @nanoporetech.com and their recent announcement that the P2 Solo will be discontinued:
rrwick.github.io/2026/01/21/p...
rrwick.github.io/2026/01/21/p...
P2 Solo announcement and the trade-offs of a more stable ONT
a blog for miscellaneous bioinformatics stuff
rrwick.github.io
January 21, 2026 at 3:38 AM
New blog post with some thoughts on @nanoporetech.com and their recent announcement that the P2 Solo will be discontinued:
rrwick.github.io/2026/01/21/p...
rrwick.github.io/2026/01/21/p...
Reposted by Jim Shaw

We just released #anvio v9, "eunice" 🎉
This version represents over 2,000 changes in the codebase since v8, increasing the total number of programs in the anvi'o ecosystem to 176.
Read the release notes:
github.com/merenlab/anv...
Visit our up-to-date web page:
anvio.org
This version represents over 2,000 changes in the codebase since v8, increasing the total number of programs in the anvi'o ecosystem to 176.
Read the release notes:
github.com/merenlab/anv...
Visit our up-to-date web page:
anvio.org

January 20, 2026 at 11:48 AM
We just released #anvio v9, "eunice" 🎉
This version represents over 2,000 changes in the codebase since v8, increasing the total number of programs in the anvi'o ecosystem to 176.
Read the release notes:
github.com/merenlab/anv...
Visit our up-to-date web page:
anvio.org
This version represents over 2,000 changes in the codebase since v8, increasing the total number of programs in the anvi'o ecosystem to 176.
Read the release notes:
github.com/merenlab/anv...
Visit our up-to-date web page:
anvio.org
Reposted by Jim Shaw

My time in @martinsteinegger.bsky.social's group is ending, but I’m staying in Korea to build a lab at Sungkyunkwan University School of Medicine. If you or someone you know is interested in molecular machine learning and open-source bioinformatics, please reach out. I am hiring!
mirdita.org
mirdita.org

Mirdita Lab - Laboratory for Computational Biology & Molecular Machine Learning
Mirdita Lab builds scalable bioinformatics methods.
mirdita.org
January 20, 2026 at 11:07 AM
My time in @martinsteinegger.bsky.social's group is ending, but I’m staying in Korea to build a lab at Sungkyunkwan University School of Medicine. If you or someone you know is interested in molecular machine learning and open-source bioinformatics, please reach out. I am hiring!
mirdita.org
mirdita.org
Reposted by Jim Shaw
I am looking for a postdoc to develop high-performance algorithms in computational genomics. Email or DM me if interested. For more information, see hlilab.github.io/vacancies. RTs appreciated!
HLi Lab - Vacancies
Openings
hlilab.github.io
January 14, 2026 at 3:44 PM
I am looking for a postdoc to develop high-performance algorithms in computational genomics. Email or DM me if interested. For more information, see hlilab.github.io/vacancies. RTs appreciated!
Reposted by Jim Shaw
Phold's manuscript is now available @narjournal.bsky.social thanks to @susiegriggo.bsky.social @npbhavya.bsky.social @vijinim.bsky.social @linsalrob.bsky.social @martinsteinegger.bsky.social @milot.bsky.social @eunbelivable.bsky.social & others not on bsky #phagesky academic.oup.com/nar/article/...
January 14, 2026 at 5:10 AM
Phold's manuscript is now available @narjournal.bsky.social thanks to @susiegriggo.bsky.social @npbhavya.bsky.social @vijinim.bsky.social @linsalrob.bsky.social @martinsteinegger.bsky.social @milot.bsky.social @eunbelivable.bsky.social & others not on bsky #phagesky academic.oup.com/nar/article/...
Reposted by Jim Shaw
Now published in Algorithms for Molecular Biology: link.springer.com/article/10.1.... Key message: a tiny CNN model with 7k parameters can capture main splice signals across vertebrates+insect and halves the minimap2 & miniprot junction error rate. I always use this new feature now.
Preprint on "Improving spliced alignment by modeling splice sites with deep learning". It describes minisplice for modeling splice signals. Minimap2 and miniprot now optionally use the predicted scores to improve spliced alignment.
arxiv.org/abs/2506.12986
arxiv.org/abs/2506.12986
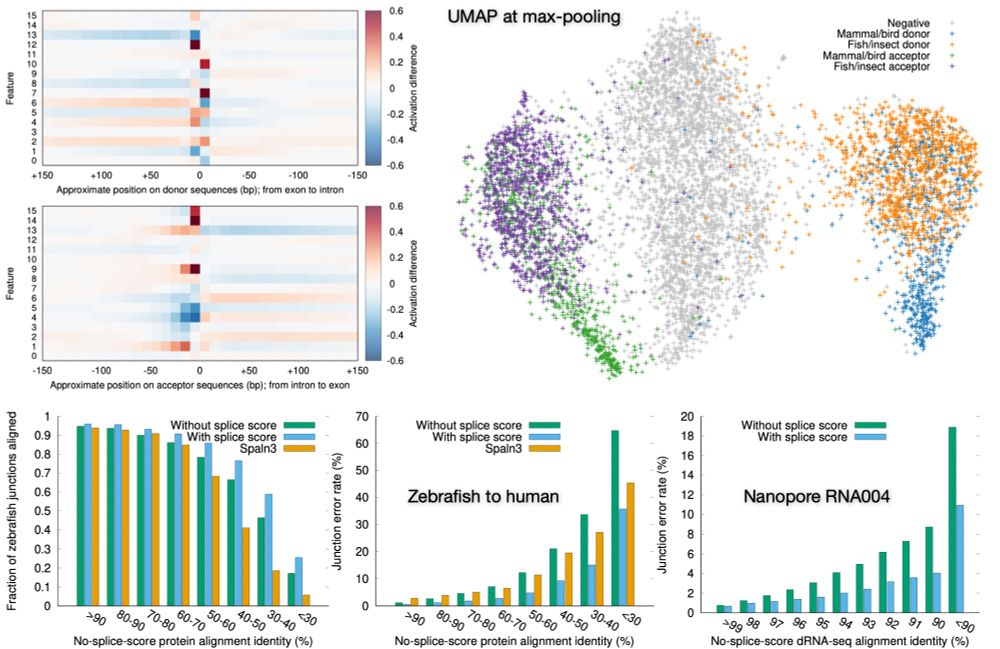
January 6, 2026 at 11:02 PM
Now published in Algorithms for Molecular Biology: link.springer.com/article/10.1.... Key message: a tiny CNN model with 7k parameters can capture main splice signals across vertebrates+insect and halves the minimap2 & miniprot junction error rate. I always use this new feature now.
Reposted by Jim Shaw

🎉 New year, NEW PREPRINT!
Bacteria exhibit astonishing genetic diversity, but where do new genes come from?
My best friend Arya Kaul (/labmate in the @baym lab) investigates how advantageous deletions can spawn new genes - "deletion-born fusions." 🧵:
Bacteria exhibit astonishing genetic diversity, but where do new genes come from?
My best friend Arya Kaul (/labmate in the @baym lab) investigates how advantageous deletions can spawn new genes - "deletion-born fusions." 🧵:

Novel genes arise from genomic deletions across the bacterial tree of life https://www.biorxiv.org/content/10.64898/2026.01.05.697752v1
January 6, 2026 at 4:09 PM
🎉 New year, NEW PREPRINT!
Bacteria exhibit astonishing genetic diversity, but where do new genes come from?
My best friend Arya Kaul (/labmate in the @baym lab) investigates how advantageous deletions can spawn new genes - "deletion-born fusions." 🧵:
Bacteria exhibit astonishing genetic diversity, but where do new genes come from?
My best friend Arya Kaul (/labmate in the @baym lab) investigates how advantageous deletions can spawn new genes - "deletion-born fusions." 🧵:
Reposted by Jim Shaw

Proud to announce SimPhyNI, a new tool for bacterial GWAS with higher precision and scalability than existing tools. Try it out and let us know what you think!!

High Precision Binary Trait Association on Phylogenetic Trees https://www.biorxiv.org/content/10.64898/2025.12.24.696407v1
January 5, 2026 at 2:55 PM
Proud to announce SimPhyNI, a new tool for bacterial GWAS with higher precision and scalability than existing tools. Try it out and let us know what you think!!
Reposted by Jim Shaw

Strobealign v0.17.0 is out! github.com/ksahlin/stro...

Release Version 0.17.0 · ksahlin/strobealign
Changes
#504: Introduce collinear chaining as the new default mapping and alignment method, replacing NAMs. This reproduces the Minimap2 chaining algorithm providing better mapping accuracy. NAMs ...
github.com
December 29, 2025 at 5:50 AM
Strobealign v0.17.0 is out! github.com/ksahlin/stro...
Reposted by Jim Shaw

Grateful to share our paper on gene-specific selective sweeps in human gut microbiomes, now out in Nature! It has been a joy to work with @rwolff.bsky.social, whose insights and hard work made this possible.
www.nature.com/articles/s41...
www.nature.com/articles/s41...

Gene-specific selective sweeps are pervasive across human gut microbiomes - Nature
Development and application of the integrated linkage disequilibrium score (iLDS) reveals both selective pressures impacting the human gut microbiome and the mechanisms by which gut bacteria adapt to ...
www.nature.com
December 17, 2025 at 6:53 PM
Grateful to share our paper on gene-specific selective sweeps in human gut microbiomes, now out in Nature! It has been a joy to work with @rwolff.bsky.social, whose insights and hard work made this possible.
www.nature.com/articles/s41...
www.nature.com/articles/s41...
Reposted by Jim Shaw

The scikit-bio paper in online in Nature Methods! Many thanks to our collaborators, community contributors and reviewers! We couldn’t have done it without you. www.nature.com/articles/s41... #Bioinformatics #OpenSource

Scikit-bio: a fundamental Python library for biological omic data analysis - Nature Methods
Nature Methods - Scikit-bio: a fundamental Python library for biological omic data analysis
www.nature.com
December 11, 2025 at 5:57 PM
The scikit-bio paper in online in Nature Methods! Many thanks to our collaborators, community contributors and reviewers! We couldn’t have done it without you. www.nature.com/articles/s41... #Bioinformatics #OpenSource
Reposted by Jim Shaw

The GTDB website now has an ANI calculator based on skani that supports uploading of user genomes. Try it at gtdb.ecogenomic.org/tools/skani.
Find more information about @jimshaw.bsky.social fantastic tool at www.nature.com/articles/s41....
Find more information about @jimshaw.bsky.social fantastic tool at www.nature.com/articles/s41....
GTDB - skani calculator
An interface to compute pairwise ANI of NCBI genomes using the GTDB taxonomy.
gtdb.ecogenomic.org
December 11, 2025 at 2:59 PM
The GTDB website now has an ANI calculator based on skani that supports uploading of user genomes. Try it at gtdb.ecogenomic.org/tools/skani.
Find more information about @jimshaw.bsky.social fantastic tool at www.nature.com/articles/s41....
Find more information about @jimshaw.bsky.social fantastic tool at www.nature.com/articles/s41....
Reposted by Jim Shaw

I’m recruiting a postdoc to work on algorithms for cancer genome reconstruction. We have access to a rich set of tumour samples sequenced across multiple technologies. If interested, feel free to DM. Please share.
December 11, 2025 at 3:04 AM
I’m recruiting a postdoc to work on algorithms for cancer genome reconstruction. We have access to a rich set of tumour samples sequenced across multiple technologies. If interested, feel free to DM. Please share.
Reposted by Jim Shaw

One flowcell from @nanoporetech.com yielded 260 Gbp 🎉🚀🤯🟩

December 8, 2025 at 4:04 PM
One flowcell from @nanoporetech.com yielded 260 Gbp 🎉🚀🤯🟩
Reposted by Jim Shaw
100%, both ONT and PacBio (although most of what we do is not marine / streamlined genome). We just published a specific study of soil metag short- vs long-read, and we see that, among other things, long-reads assemble regions too complex for short reads academic.oup.com/nargab/artic...

Comparison of short-read and long-read metagenome assemblies in a natural soil community highlights systematic bias in recovery of high-diversity populations
Abstract. Comparisons of long-read and short-read (meta)genome assemblies typically show that short-read sequence assemblies are less error-prone, but stru
academic.oup.com
December 8, 2025 at 4:25 PM
100%, both ONT and PacBio (although most of what we do is not marine / streamlined genome). We just published a specific study of soil metag short- vs long-read, and we see that, among other things, long-reads assemble regions too complex for short reads academic.oup.com/nargab/artic...