



Tom Stanton
@tomstantonmicro.bsky.social
Microbiologist. Lead developer of #Kaptive + bonafide #Klebsiella nerd. Post-doc in the Wyres Lab @AlfredMonash_ID.
Reposted by Tom Stanton

Legit work in progress photo…
www.imperial.ac.uk/news/271753/...
www.imperial.ac.uk/news/271753/...

GSK and Fleming Initiative scientists unite to target AMR with advanced AI | Imperial News | Imperial College London
£45m in GSK funding has been allocated to new research programmes combining expertise and using cutting edge AI technology to accelerate AMR research.
www.imperial.ac.uk
November 18, 2025 at 9:57 AM
Legit work in progress photo…
www.imperial.ac.uk/news/271753/...
www.imperial.ac.uk/news/271753/...
Reposted by Tom Stanton

Hard to believe it’s already that time of year!
Our kick-off meeting for the Klebsiella Seminar Series just wrapped up. Stay tuned for more!
@lauraamike.bsky.social @olayarendueles.bsky.social @caityholmes.bsky.social @tomstantonmicro.bsky.social @juanvalenciabacca.bsky.social WenWen Low & Jay V.
Our kick-off meeting for the Klebsiella Seminar Series just wrapped up. Stay tuned for more!
@lauraamike.bsky.social @olayarendueles.bsky.social @caityholmes.bsky.social @tomstantonmicro.bsky.social @juanvalenciabacca.bsky.social WenWen Low & Jay V.
November 18, 2025 at 10:43 PM
Hard to believe it’s already that time of year!
Our kick-off meeting for the Klebsiella Seminar Series just wrapped up. Stay tuned for more!
@lauraamike.bsky.social @olayarendueles.bsky.social @caityholmes.bsky.social @tomstantonmicro.bsky.social @juanvalenciabacca.bsky.social WenWen Low & Jay V.
Our kick-off meeting for the Klebsiella Seminar Series just wrapped up. Stay tuned for more!
@lauraamike.bsky.social @olayarendueles.bsky.social @caityholmes.bsky.social @tomstantonmicro.bsky.social @juanvalenciabacca.bsky.social WenWen Low & Jay V.
Reposted by Tom Stanton

The @klebnet.bsky.social team are pleased to share slides from our “Klebsiella pneumoniae Genomic Epidemiology & Antimicrobial Resistance” lecture series!
Topics include Kleb diversity, lineages, AMR, hypervirulence, how to use Kaptive & Kleborate for typing, and more!
klebnet.org/2025/11/18/k...
Topics include Kleb diversity, lineages, AMR, hypervirulence, how to use Kaptive & Kleborate for typing, and more!
klebnet.org/2025/11/18/k...
Klebsiella pneumoniae genomics tutorials – KlebNET-GSP
klebnet.org
November 18, 2025 at 8:25 AM
The @klebnet.bsky.social team are pleased to share slides from our “Klebsiella pneumoniae Genomic Epidemiology & Antimicrobial Resistance” lecture series!
Topics include Kleb diversity, lineages, AMR, hypervirulence, how to use Kaptive & Kleborate for typing, and more!
klebnet.org/2025/11/18/k...
Topics include Kleb diversity, lineages, AMR, hypervirulence, how to use Kaptive & Kleborate for typing, and more!
klebnet.org/2025/11/18/k...
Reposted by Tom Stanton

BIGSdb v1.51.4 has been released. This adds a new #Kaptive plugin for surface polysaccharide typing of Acinetobacter baumannii and Klebsiella. github.com/kjolley/BIGS... for details. Kaptive is developed by @tomstantonmicro.bsky.social, @kelwyres.bsky.social , @katholt.bsky.social and colleagues.

July 18, 2025 at 7:57 AM
BIGSdb v1.51.4 has been released. This adds a new #Kaptive plugin for surface polysaccharide typing of Acinetobacter baumannii and Klebsiella. github.com/kjolley/BIGS... for details. Kaptive is developed by @tomstantonmicro.bsky.social, @kelwyres.bsky.social , @katholt.bsky.social and colleagues.
Reposted by Tom Stanton

Our new #klebsiella O type nomenclature, codesigned with Chris Whitfield, is now live in @pathogenwatch.bsky.social!
Need a quick explainer on the new names? Check out my blog post: tinyurl.com/y8yb3rbb (+link to full review article)
#MicroSky @klebnet.bsky.social @tomstantonmicro.bsky.social
Need a quick explainer on the new names? Check out my blog post: tinyurl.com/y8yb3rbb (+link to full review article)
#MicroSky @klebnet.bsky.social @tomstantonmicro.bsky.social
July 22, 2025 at 5:18 AM
Our new #klebsiella O type nomenclature, codesigned with Chris Whitfield, is now live in @pathogenwatch.bsky.social!
Need a quick explainer on the new names? Check out my blog post: tinyurl.com/y8yb3rbb (+link to full review article)
#MicroSky @klebnet.bsky.social @tomstantonmicro.bsky.social
Need a quick explainer on the new names? Check out my blog post: tinyurl.com/y8yb3rbb (+link to full review article)
#MicroSky @klebnet.bsky.social @tomstantonmicro.bsky.social
Reposted by Tom Stanton

We are pleased to launch the KlebNET Genomic Epidemiology Consortium!
We aim to build a public metadata repository; systematic risk framework for global genomic surveillance; and genomic epi reviews for high-impact #Klebsiella clones.
Join us here:
klebnet.org/klebnet-gsp-...
#ABPHM25
We aim to build a public metadata repository; systematic risk framework for global genomic surveillance; and genomic epi reviews for high-impact #Klebsiella clones.
Join us here:
klebnet.org/klebnet-gsp-...
#ABPHM25
KlebNET-GSP
klebnet.org
May 21, 2025 at 12:40 PM
We are pleased to launch the KlebNET Genomic Epidemiology Consortium!
We aim to build a public metadata repository; systematic risk framework for global genomic surveillance; and genomic epi reviews for high-impact #Klebsiella clones.
Join us here:
klebnet.org/klebnet-gsp-...
#ABPHM25
We aim to build a public metadata repository; systematic risk framework for global genomic surveillance; and genomic epi reviews for high-impact #Klebsiella clones.
Join us here:
klebnet.org/klebnet-gsp-...
#ABPHM25
Reposted by Tom Stanton

Always a pleasure to organize this with @caityholmes.bsky.social @lauraamike.bsky.social Jay Vornhagen, Wen wen low, & this year joining us @tomstantonmicro.bsky.social & Juan Valencia.
April 1, 2025 at 8:30 AM
Always a pleasure to organize this with @caityholmes.bsky.social @lauraamike.bsky.social Jay Vornhagen, Wen wen low, & this year joining us @tomstantonmicro.bsky.social & Juan Valencia.
Janitor saves the day again!
www.rdocumentation.org/packages/jan...
www.rdocumentation.org/packages/jan...
excel_numeric_to_date function - RDocumentation
<p>Converts numbers like <code>42370</code> into date values like
<code>2016-01-01</code>.</p>
<p>Defaults to the modern Excel date encoding system. However, Excel for Mac
2008 and earlier Mac version...
www.rdocumentation.org
February 19, 2025 at 4:05 AM
Janitor saves the day again!
www.rdocumentation.org/packages/jan...
www.rdocumentation.org/packages/jan...
Reposted by Tom Stanton

K. variicola can be misID as K. pneumoniae. Our #OpenAccess paper reports an NICU #Outbreak
📌The rapid detection of a neonatal unit outbreak of a wild-type Klebsiella variicola using decentralized Oxford Nanopore sequencing
doi.org/10.1186/s137...
@nanoporetech.com
🖥️🧬💻
#AcademicSky
#Microsky
🧪🧫🦠
📌The rapid detection of a neonatal unit outbreak of a wild-type Klebsiella variicola using decentralized Oxford Nanopore sequencing
doi.org/10.1186/s137...
@nanoporetech.com
🖥️🧬💻
#AcademicSky
#Microsky
🧪🧫🦠

The rapid detection of a neonatal unit outbreak of a wild-type Klebsiella variicola using decentralized Oxford Nanopore sequencing - Antimicrobial Resistance & Infection Control
Background Klebsiella variicola has been implicated in neonatal intensive care unit (NICU) outbreaks previously and can be misidentified as Klebsiella pneumoniae. An increased incidence of K. pneumoni...
doi.org
February 11, 2025 at 3:13 AM
K. variicola can be misID as K. pneumoniae. Our #OpenAccess paper reports an NICU #Outbreak
📌The rapid detection of a neonatal unit outbreak of a wild-type Klebsiella variicola using decentralized Oxford Nanopore sequencing
doi.org/10.1186/s137...
@nanoporetech.com
🖥️🧬💻
#AcademicSky
#Microsky
🧪🧫🦠
📌The rapid detection of a neonatal unit outbreak of a wild-type Klebsiella variicola using decentralized Oxford Nanopore sequencing
doi.org/10.1186/s137...
@nanoporetech.com
🖥️🧬💻
#AcademicSky
#Microsky
🧪🧫🦠
Reposted by Tom Stanton

New pre print! We establish an ex vivo blood vessel model to investigate the effect of infection on vascular physiology in real time. We show Klebsiella inhibits vasodilation in a T6SS-dependent manner by targeting eNOs. Superb work by @safimicro.bsky.social www.biorxiv.org/content/10.1...

Klebsiella pneumoniae disrupts vasodilation by targeting eNOS post translational modifications via the type VI secretion system and the capsule polysaccharide
Vasodilation is a crucial protective response to inflammation and infection. Endothelial cells control vasodilation through the bioavailability of eNOS-produced nitric oxide (NO), and the generation o...
www.biorxiv.org
February 6, 2025 at 9:15 AM
New pre print! We establish an ex vivo blood vessel model to investigate the effect of infection on vascular physiology in real time. We show Klebsiella inhibits vasodilation in a T6SS-dependent manner by targeting eNOs. Superb work by @safimicro.bsky.social www.biorxiv.org/content/10.1...
Lastly, we'd like to thank YOU, the Kaptive community, for guiding development, spotting bugs and collaborating with us!
But this is just the beginning, we have lots of exciting things in store for the future of Kaptive to make in silico serotyping even better!
#kaptive #klebsiella #acinetobacter
But this is just the beginning, we have lots of exciting things in store for the future of Kaptive to make in silico serotyping even better!
#kaptive #klebsiella #acinetobacter
February 9, 2025 at 3:19 AM
Lastly, we'd like to thank YOU, the Kaptive community, for guiding development, spotting bugs and collaborating with us!
But this is just the beginning, we have lots of exciting things in store for the future of Kaptive to make in silico serotyping even better!
#kaptive #klebsiella #acinetobacter
But this is just the beginning, we have lots of exciting things in store for the future of Kaptive to make in silico serotyping even better!
#kaptive #klebsiella #acinetobacter
Kaptive 3 is now integrated within Kaptive-Web (kaptive-web.erc.monash.edu), PathogenWatch (pathogen.watch), the new Kleborate 3 framework (github.com/klebgenomics...) and Bactopia (bactopia.github.io/latest/).
Remember to cite us if you use Kaptive for your results, and watch out for "Untypeable"!
Remember to cite us if you use Kaptive for your results, and watch out for "Untypeable"!
Pathogenwatch
A global platform for genomic surveillance.
pathogen.watch
February 9, 2025 at 3:19 AM
Kaptive 3 is now integrated within Kaptive-Web (kaptive-web.erc.monash.edu), PathogenWatch (pathogen.watch), the new Kleborate 3 framework (github.com/klebgenomics...) and Bactopia (bactopia.github.io/latest/).
Remember to cite us if you use Kaptive for your results, and watch out for "Untypeable"!
Remember to cite us if you use Kaptive for your results, and watch out for "Untypeable"!
We know the command-line can be tricky, so we made the CLI much friendlier 🧑💻
For the code-savvy, there's also a Python API allowing Kaptive to be used within your own programs 🧱
All the information you need is in the documentation, which we update regularly: kaptive.readthedocs.io/en/latest/
For the code-savvy, there's also a Python API allowing Kaptive to be used within your own programs 🧱
All the information you need is in the documentation, which we update regularly: kaptive.readthedocs.io/en/latest/
Introducing Kaptive 3 — Kaptive 3.0.0 documentation
kaptive.readthedocs.io
February 9, 2025 at 3:19 AM
We know the command-line can be tricky, so we made the CLI much friendlier 🧑💻
For the code-savvy, there's also a Python API allowing Kaptive to be used within your own programs 🧱
All the information you need is in the documentation, which we update regularly: kaptive.readthedocs.io/en/latest/
For the code-savvy, there's also a Python API allowing Kaptive to be used within your own programs 🧱
All the information you need is in the documentation, which we update regularly: kaptive.readthedocs.io/en/latest/
Kaptive 3 is also much (much) faster than Kaptive 2, taking ~1 second per assembly 🏎️💨
This means that if you don't have a fancy HPC, then don't worry! You can still analyse thousands of your own assemblies on your laptop in a reasonable time! 💻
This means that if you don't have a fancy HPC, then don't worry! You can still analyse thousands of your own assemblies on your laptop in a reasonable time! 💻

February 9, 2025 at 3:19 AM
Kaptive 3 is also much (much) faster than Kaptive 2, taking ~1 second per assembly 🏎️💨
This means that if you don't have a fancy HPC, then don't worry! You can still analyse thousands of your own assemblies on your laptop in a reasonable time! 💻
This means that if you don't have a fancy HPC, then don't worry! You can still analyse thousands of your own assemblies on your laptop in a reasonable time! 💻
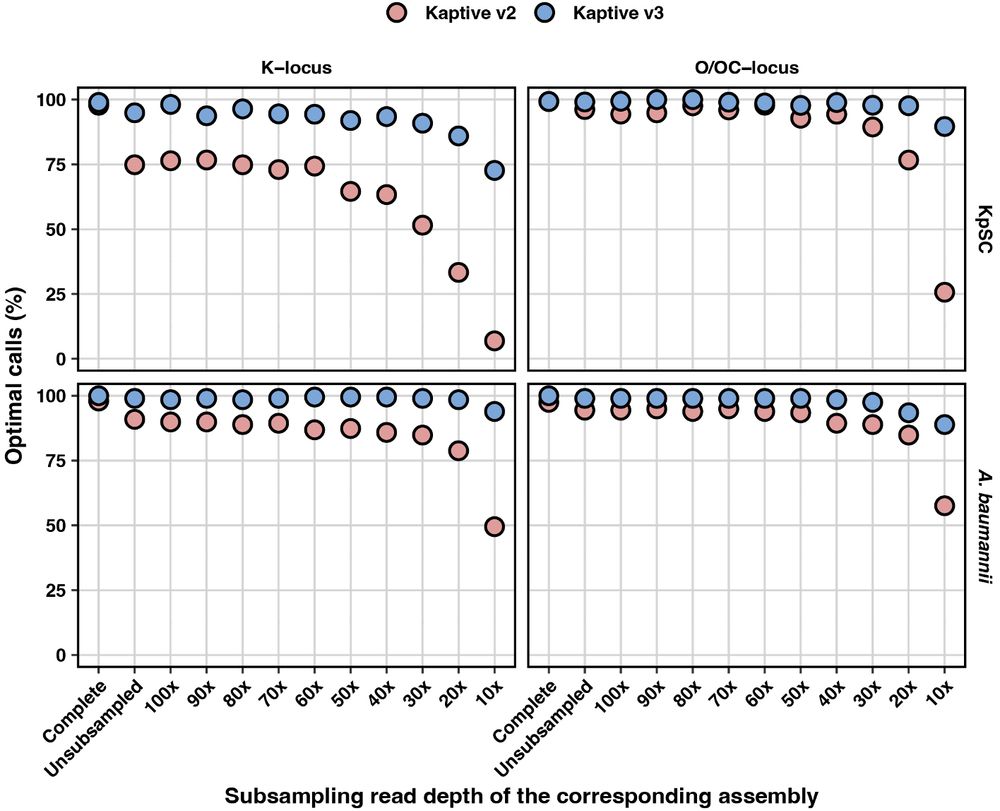
We then subsampled the corresponding short reads at decreasing depths and created sets of increasingly awful draft assemblies with loci broken over contigs and lots of genes missing.
Kaptive 3 was much more sensitive than Kaptive 2, and maintained accuracy even when the assemblies were awful! 💩
Kaptive 3 was much more sensitive than Kaptive 2, and maintained accuracy even when the assemblies were awful! 💩
February 9, 2025 at 3:19 AM
We then subsampled the corresponding short reads at decreasing depths and created sets of increasingly awful draft assemblies with loci broken over contigs and lots of genes missing.
Kaptive 3 was much more sensitive than Kaptive 2, and maintained accuracy even when the assemblies were awful! 💩
Kaptive 3 was much more sensitive than Kaptive 2, and maintained accuracy even when the assemblies were awful! 💩
We put together a special dataset specifically designed to test Kaptive. We had completed hybrid assemblies for KpSC (preprints.scielo.org/index.php/sc...) and A. baumannii (RefSeq).
We identified the K- and O(C)-loci in each and visually confirmed each to determine a ground truth Kaptive call 🔎
We identified the K- and O(C)-loci in each and visually confirmed each to determine a ground truth Kaptive call 🔎
Complete genomes of 568 diverse Klebsiella pneumoniae species complex isolates from humans, animals and marine sources in Norway from 2001-2020
We report 579 hybrid genome assemblies (568 complete) of Klebsiella pneumoniae species complex isolates from human, animal and marine sources in Norway collected 2001-2020, belonging to six phylogroups including K. pneumoniae (n=493) and K. variicola (n=69) and 364 unique sequence types.
preprints.scielo.org
February 9, 2025 at 3:19 AM
We put together a special dataset specifically designed to test Kaptive. We had completed hybrid assemblies for KpSC (preprints.scielo.org/index.php/sc...) and A. baumannii (RefSeq).
We identified the K- and O(C)-loci in each and visually confirmed each to determine a ground truth Kaptive call 🔎
We identified the K- and O(C)-loci in each and visually confirmed each to determine a ground truth Kaptive call 🔎
So enter Kaptive 3, a complete overhaul of Kaptive with a new algorithm designed to handle fragmented loci.
We also refactored (and simplified) the confidence score to be more sensitive for broken loci and missing genes, allowing more Kaptive data to be used when the assembly may not be complete 💯
We also refactored (and simplified) the confidence score to be more sensitive for broken loci and missing genes, allowing more Kaptive data to be used when the assembly may not be complete 💯

February 9, 2025 at 3:19 AM
So enter Kaptive 3, a complete overhaul of Kaptive with a new algorithm designed to handle fragmented loci.
We also refactored (and simplified) the confidence score to be more sensitive for broken loci and missing genes, allowing more Kaptive data to be used when the assembly may not be complete 💯
We also refactored (and simplified) the confidence score to be more sensitive for broken loci and missing genes, allowing more Kaptive data to be used when the assembly may not be complete 💯
Because of how Kaptive 2 chose the best match locus, missing locus sequence resulted in a coverage bias for shorter loci in the database such, and could sometimes lead to inaccurate calls!
Ever seen a stray KL107 in your data that didn't make sense?
Yeah, that's why...
Ever seen a stray KL107 in your data that didn't make sense?
Yeah, that's why...
February 9, 2025 at 3:19 AM
Because of how Kaptive 2 chose the best match locus, missing locus sequence resulted in a coverage bias for shorter loci in the database such, and could sometimes lead to inaccurate calls!
Ever seen a stray KL107 in your data that didn't make sense?
Yeah, that's why...
Ever seen a stray KL107 in your data that didn't make sense?
Yeah, that's why...
So in a nutshell, we traced Kaptive's issues with the Klebsiella K-locus all the way back to the gDNA, where:
The locus region is partially amplified ->
Low sequencing read coverage ->
region doesn't assemble well ->
Untypeable Kaptive call ->
Unusable data 🙅♀️
The locus region is partially amplified ->
Low sequencing read coverage ->
region doesn't assemble well ->
Untypeable Kaptive call ->
Unusable data 🙅♀️
February 9, 2025 at 3:19 AM
So in a nutshell, we traced Kaptive's issues with the Klebsiella K-locus all the way back to the gDNA, where:
The locus region is partially amplified ->
Low sequencing read coverage ->
region doesn't assemble well ->
Untypeable Kaptive call ->
Unusable data 🙅♀️
The locus region is partially amplified ->
Low sequencing read coverage ->
region doesn't assemble well ->
Untypeable Kaptive call ->
Unusable data 🙅♀️
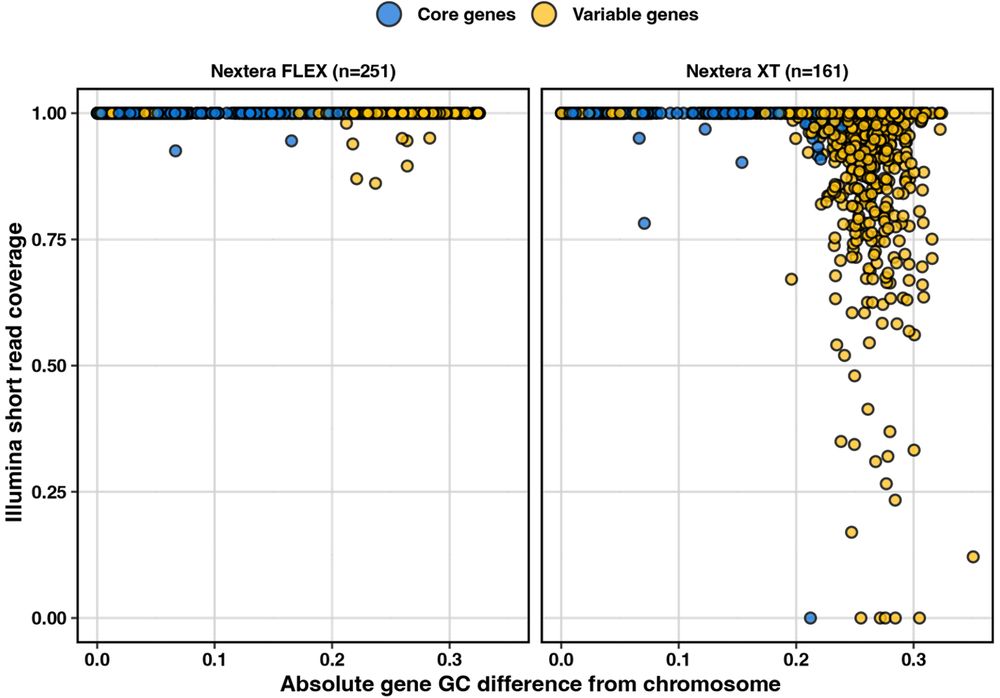
These genes have a very low GC compared to the rest of the Klebsiella chromosome, so we wondered if this was affecting how this part of the genome gets sequenced.
Turns out, these genes show decreased sequencing coverage when reads are prepped with Nextera XT, but not so much with Nextera Flex 🤯
Turns out, these genes show decreased sequencing coverage when reads are prepped with Nextera XT, but not so much with Nextera Flex 🤯
February 9, 2025 at 3:19 AM
These genes have a very low GC compared to the rest of the Klebsiella chromosome, so we wondered if this was affecting how this part of the genome gets sequenced.
Turns out, these genes show decreased sequencing coverage when reads are prepped with Nextera XT, but not so much with Nextera Flex 🤯
Turns out, these genes show decreased sequencing coverage when reads are prepped with Nextera XT, but not so much with Nextera Flex 🤯
The major drivers of low confidence were K-loci that were 1) broken over contigs 🚫 and 2) missing genes ➡️➡️, events that were mostly co-occurring
Turns out, the genes missing were usually those important for antigenic diversity, in this case the glycosyltransferases that dictate the CPS 🍬 structure
Turns out, the genes missing were usually those important for antigenic diversity, in this case the glycosyltransferases that dictate the CPS 🍬 structure
February 9, 2025 at 3:19 AM
The major drivers of low confidence were K-loci that were 1) broken over contigs 🚫 and 2) missing genes ➡️➡️, events that were mostly co-occurring
Turns out, the genes missing were usually those important for antigenic diversity, in this case the glycosyltransferases that dictate the CPS 🍬 structure
Turns out, the genes missing were usually those important for antigenic diversity, in this case the glycosyltransferases that dictate the CPS 🍬 structure
So you may have noticed your Klebsiella K-locus results from previous versions of Kaptive (v1-2) having lots of untypeable calls ("Low" + "None" confidence) with draft assemblies; we certainly did!
This meant that lots of useful seroepi data was unusable, so we started by finding out exactly why 🤔
This meant that lots of useful seroepi data was unusable, so we started by finding out exactly why 🤔
February 9, 2025 at 3:19 AM
So you may have noticed your Klebsiella K-locus results from previous versions of Kaptive (v1-2) having lots of untypeable calls ("Low" + "None" confidence) with draft assemblies; we certainly did!
This meant that lots of useful seroepi data was unusable, so we started by finding out exactly why 🤔
This meant that lots of useful seroepi data was unusable, so we started by finding out exactly why 🤔
Super excited to finally present the preprint to accompany Kaptive 3 which we released last year!
Big thanks to coauthors @kelwyres.bsky.social, @katholt.bsky.social, @genomarit.bsky.social and Iren Löhr.
Here's what we did to improve in silico antigen typing 👇🧵
www.biorxiv.org/content/10.1...
Big thanks to coauthors @kelwyres.bsky.social, @katholt.bsky.social, @genomarit.bsky.social and Iren Löhr.
Here's what we did to improve in silico antigen typing 👇🧵
www.biorxiv.org/content/10.1...

Fast and Accurate in silico Antigen Typing with Kaptive 3
Surface polysaccharides are common antigens in priority pathogens and therefore attractive targets for novel control strategies such as vaccines, monoclonal antibody and phage therapies. Distinct serotypes correspond to diverse polysaccharide structures that are encoded by distinct biosynthesis gene clusters, e.g. the Klebsiella pneumoniae species complex (KpSC) K- and O- loci encode the synthesis machinery for the capsule (K) and outer-lipopolysaccharides (O), respectively. We previously presented Kaptive and Kaptive 2, programs to identify K and O-loci directly from KpSC genome assemblies (later adapted for Acinetobacter baumannii), enabling sero-epidemiological analyses to guide vaccine and phage therapy development. However, for some KpSC genome collections, Kaptive (v≤2) was unable to type a high proportion of K-loci. Here we identify the cause of this issue as assembly fragmentation, and present a new version of Kaptive (v3) to circumvent this problem, reduce processing times and simplify output interpretation. We compared the performance of Kaptive v2 and Kaptive v3 for typing genome assemblies generated from subsampled Illumina read sets (decrements of 10x depth), for which a corresponding high quality completed genome was also available to determine the 'true' loci (n=549 KpSC, n=198 A. baumannii). Both versions of Kaptive showed high rates of agreement to the matched true locus among 'typeable' locus calls (≥96% for ≥20x read depth), but Kaptive v3 was more sensitive, particularly for low depth assemblies (at <40x depth, v3 ranged 0.85-1 vs v2 0.09-0.94) and/or typing KpSC K-loci (e.g. 0.97 vs 0.82 for non-subsampled assemblies). Overall, Kaptive v3 was also associated with a higher rate of optimal outcomes i.e. loci matching those in the reference database were correctly typed and genuine novel loci were reported as untypeable (73-98% for v3 vs 7-77% for v2 for KpSC K-loci). Kaptive v3 was >1 order of magnitude faster than Kaptive v2 making it easy to analyse thousands of assemblies on a desktop computer, facilitating broadly accessible in silico serotyping that is both accurate and sensitive. The Kaptive v3 source code is freely available on GitHub (https://github.com/klebgenomics/Kaptive), and has been implemented in Kaptive Web (https://kaptive-web.erc.monash.edu). ### Competing Interest Statement The authors have declared no competing interest.
www.biorxiv.org
February 9, 2025 at 3:19 AM
Super excited to finally present the preprint to accompany Kaptive 3 which we released last year!
Big thanks to coauthors @kelwyres.bsky.social, @katholt.bsky.social, @genomarit.bsky.social and Iren Löhr.
Here's what we did to improve in silico antigen typing 👇🧵
www.biorxiv.org/content/10.1...
Big thanks to coauthors @kelwyres.bsky.social, @katholt.bsky.social, @genomarit.bsky.social and Iren Löhr.
Here's what we did to improve in silico antigen typing 👇🧵
www.biorxiv.org/content/10.1...
Reposted by Tom Stanton

The PopPIPE (github.com/bacpop/PopPIPE) analysis pipeline can be used to subcluster data, create visualisations and run transmission analyses.
Preprint now here: www.biorxiv.org/content/10.1...
Including a case study on nosocomial transmission of vancomycin resistant Enterococcus faecium
Preprint now here: www.biorxiv.org/content/10.1...
Including a case study on nosocomial transmission of vancomycin resistant Enterococcus faecium
GitHub - bacpop/PopPIPE: Population analysis PIPEline 🛠🧬
Population analysis PIPEline 🛠🧬. Contribute to bacpop/PopPIPE development by creating an account on GitHub.
github.com
December 10, 2024 at 9:52 AM
The PopPIPE (github.com/bacpop/PopPIPE) analysis pipeline can be used to subcluster data, create visualisations and run transmission analyses.
Preprint now here: www.biorxiv.org/content/10.1...
Including a case study on nosocomial transmission of vancomycin resistant Enterococcus faecium
Preprint now here: www.biorxiv.org/content/10.1...
Including a case study on nosocomial transmission of vancomycin resistant Enterococcus faecium
