
Marcel M
@mrclmllr.bsky.social
200 followers
190 following
32 posts
Postdoctoral Fellow at @thematterlab.bsky.social with @aspuru.bsky.social | PhD in Theoretical Chemistry | ex FCI scholar & Digital Chemistry @merckgroup.bsky.social
Posts
Media
Videos
Starter Packs
Reposted by Marcel M

Grimme Lab
@grimmelab.bsky.social
· Sep 3

Chemical Space Exploration with Artificial “Mindless” Molecules
We introduce MindlessGen, a Python-based generator for creating chemically diverse, “mindless” molecules through random atomic placement and subsequent geometry optimization. Using this framework, we ...
doi.org
Reposted by Marcel M


Reposted by Marcel M


Reposted by Marcel M


Marcel M
@mrclmllr.bsky.social
· Jun 24
Marcel M
@mrclmllr.bsky.social
· Jun 24
Marcel M
@mrclmllr.bsky.social
· Jun 24
Marcel M
@mrclmllr.bsky.social
· Jun 24

GitHub - grimme-lab/g-xtb: Development versions of the g-xTB method. Final implementation will not happen here but in tblite (https://github.com/tblite/tblite).
Development versions of the g-xTB method. Final implementation will not happen here but in tblite (https://github.com/tblite/tblite). - grimme-lab/g-xtb
github.com
Marcel M
@mrclmllr.bsky.social
· Jun 24
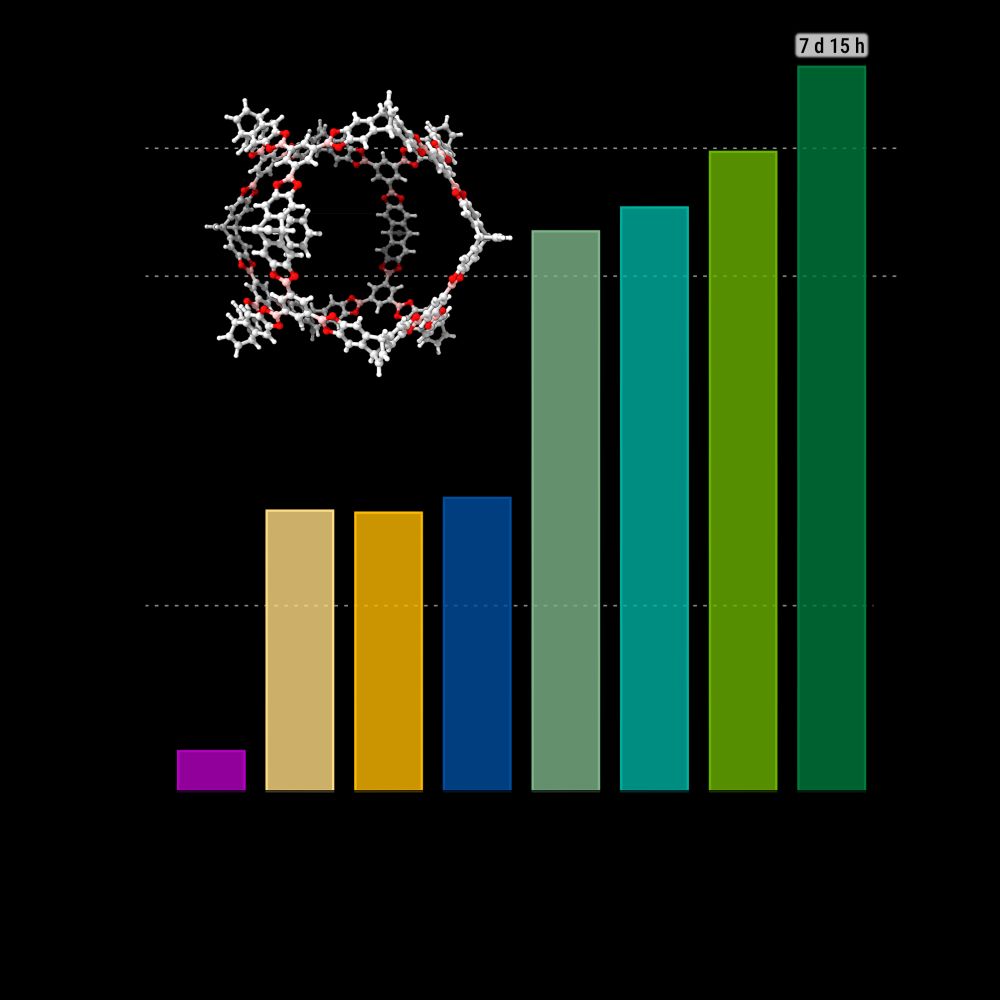
Marcel M
@mrclmllr.bsky.social
· Jun 24

Chemical Space Exploration with Artificial ”Mindless” Molecules
We introduce MindlessGen, a Python-based generator for creating chemically diverse, “mindless” molecules through random atomic placement and subsequent geometry optimization. Using this framework, we ...
chemrxiv.org

Marcel M
@mrclmllr.bsky.social
· Jun 24
Marcel M
@mrclmllr.bsky.social
· Jun 24
Marcel M
@mrclmllr.bsky.social
· Jun 24

g-xTB: A General-Purpose Extended Tight-Binding Electronic Structure Method For the Elements H to Lr (Z=1–103)
We present g-xTB, a next-generation semi-empirical electronic structure method derived from tight-binding (TB) approximations to Kohn–Sham density functional theory (KS-DFT). Designed to bridge the ga...
chemrxiv.org
Marcel M
@mrclmllr.bsky.social
· Jun 23
Marcel M
@mrclmllr.bsky.social
· Jun 23




Marcel M
@mrclmllr.bsky.social
· Feb 14
Reposted by Marcel M

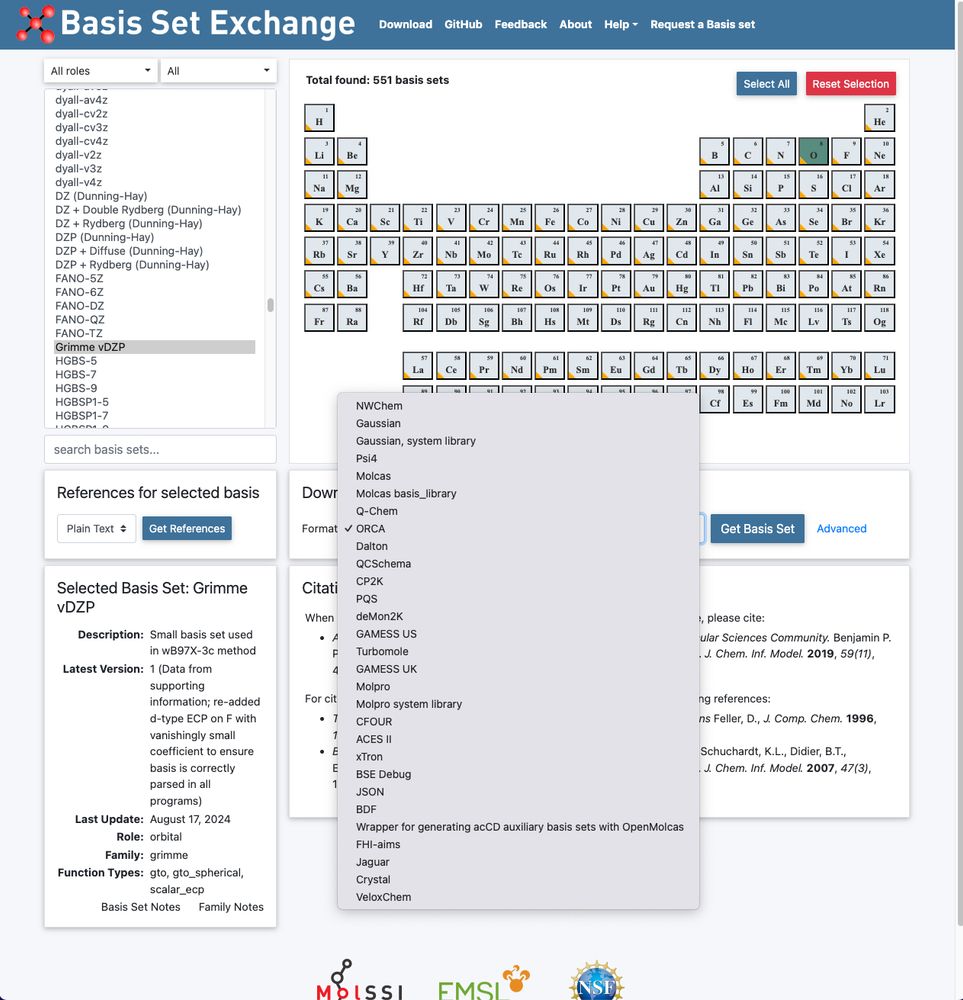
Marcel M
@mrclmllr.bsky.social
· Jan 23