
Grimme Lab
@grimmelab.bsky.social
1.7K followers
270 following
14 posts
Development and application of efficient computational chemistry methods - based @unibonn.bsky.social.
This account is managed by group members of Prof. Stefan Grimme.
Posts
Media
Videos
Starter Packs




Reposted by Grimme Lab

Johannes Gorges
@jogorges.bsky.social
· Sep 8
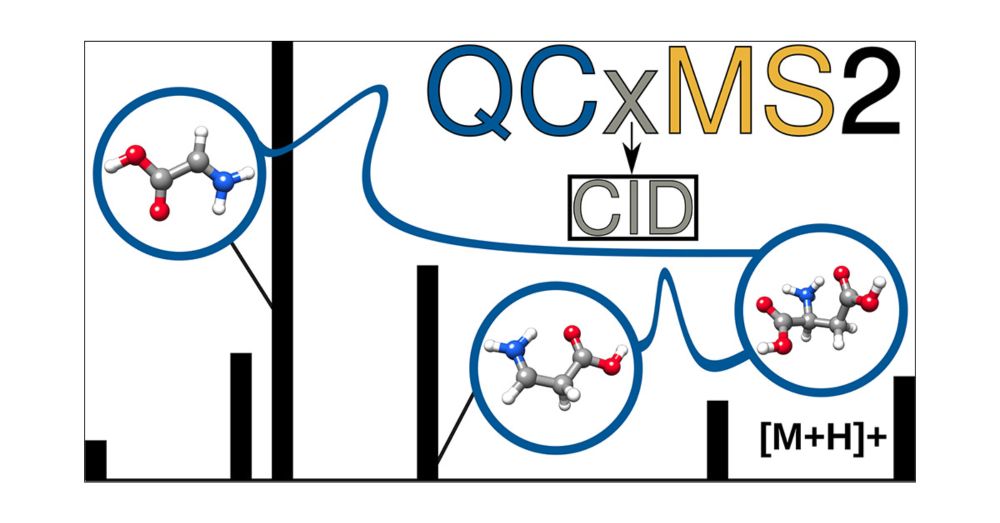
Evaluation of the QCxMS2 Method for the Calculation of Collision-Induced Dissociation Spectra via Automated Reaction Network Exploration
Collision-induced dissociation mass spectrometry (CID-MS) is an important tool in analytical chemistry for the structural elucidation of unknown compounds. The theoretical prediction of the CID spectra plays a critical role in supporting and accelerating this process. To this end, we adapt the recently developed QCxMS2 program originally designed for the calculation of electron ionization (EI) spectra to enable the computation of CID-MS. To account for the fragmentation conditions characteristic of CID within the automated reaction network discovery approach of QCxMS2 we adapted the internal energy distribution to match the experimental conditions. This distribution can be adjusted via a single parameter to approximate various activation settings, thereby eliminating the need for explicit simulations of the collisional process. We evaluate our approach on a test set of 13 organic molecules with diverse functional groups, compiled specifically for this study. All reference spectra were recorded consistently under the same measurement conditions, including both CID and higher-energy collisional dissociation (HCD) modes. Overall, QCxMS2 achieves a good average entropy similarity score (ESS) of 0.687 for the HCD spectra and 0.773 for the CID spectra. The direct comparison to experimental data demonstrates that the QCxMS2 approach, even without explicit modeling of collisions, is generally capable of computing both CID and HCD spectra with reasonable accuracy and robustness. This highlights its potential as a valuable tool for integration into structure elucidation workflows in analytical mass spectrometry.
doi.org

Grimme Lab
@grimmelab.bsky.social
· Sep 3

Chemical Space Exploration with Artificial “Mindless” Molecules
We introduce MindlessGen, a Python-based generator for creating chemically diverse, “mindless” molecules through random atomic placement and subsequent geometry optimization. Using this framework, we ...
doi.org
Reposted by Grimme Lab

Reposted by Grimme Lab
Reposted by Grimme Lab

Jan Řezáč
@jrezac.bsky.social
· Jun 26
Reposted by Grimme Lab

Marcel M
@mrclmllr.bsky.social
· Jun 24

g-xTB: A General-Purpose Extended Tight-Binding Electronic Structure Method For the Elements H to Lr (Z=1–103)
We present g-xTB, a next-generation semi-empirical electronic structure method derived from tight-binding (TB) approximations to Kohn–Sham density functional theory (KS-DFT). Designed to bridge the ga...
chemrxiv.org


Grimme Lab
@grimmelab.bsky.social
· Apr 7
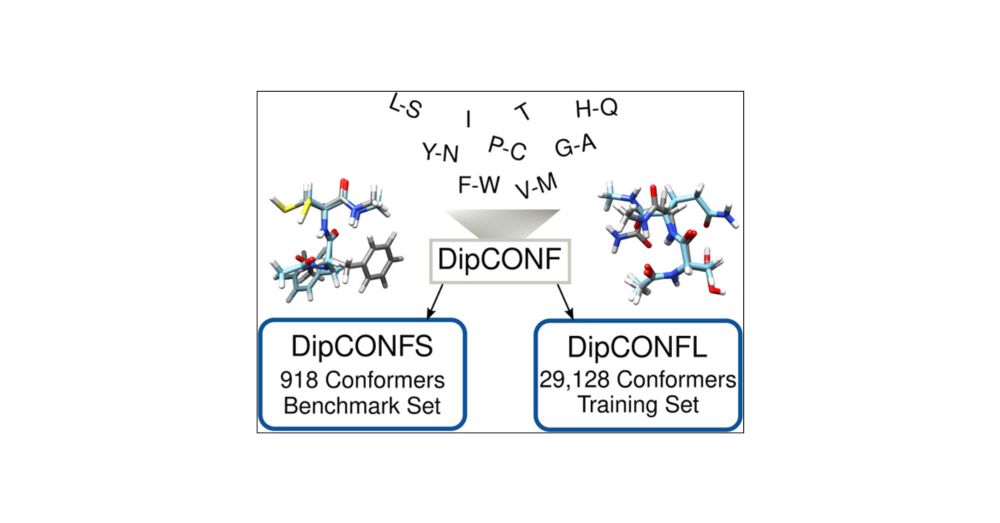
Toward Reliable Conformational Energies of Amino Acids and Dipeptides─The DipCONFS Benchmark and DipCONL Datasets
Simulating peptides and proteins is becoming increasingly important, leading to a growing need for efficient computational methods. These are typically semiempirical quantum mechanical (SQM) methods, ...
pubs.acs.org
Reposted by Grimme Lab

Lisa Pecher
@lisipea.bsky.social
· Apr 5
Best‐Practice DFT Protocols for Basic Molecular Computational Chemistry**
Many chemical investigations are supported by routine calculations of molecular structures, reaction energies, barrier heights, and spectroscopic properties. Most of these quantum-chemical calculatio...
onlinelibrary.wiley.com

Reposted by Grimme Lab



Reposted by Grimme Lab
Johannes Gorges
@jogorges.bsky.social
· Mar 7
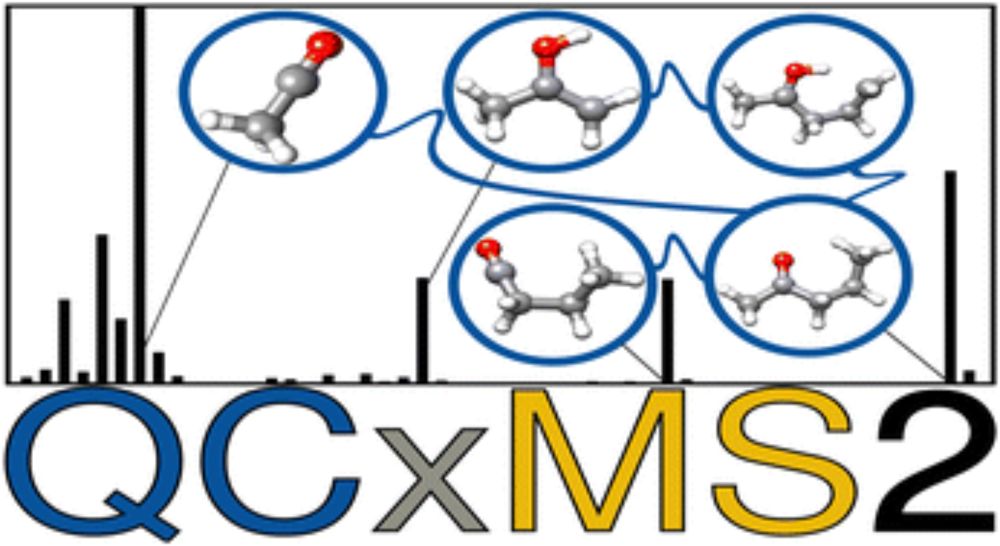
QCxMS2 – a program for the calculation of electron ionization mass spectra via automated reaction network discovery
We present a new fully-automated computational workflow for the calculation of electron ionization mass spectra by automated reaction network discovery, transition state theory and Monte-Carlo simulat...
pubs.rsc.org


Reposted by Grimme Lab

FACCTs
@faccts.de
· Feb 17

QCxMS2 - a program for the calculation of electron ion- ization mass spectra via automated reaction network dis- covery
We present a new fully-automated computational workflow for the calculation of electron ionization mass spectra by automated reaction network discovery, transition state theory and Monte-Carlo simulat...
doi.org
Grimme Lab
@grimmelab.bsky.social
· Feb 5
Johannes Gorges
@jogorges.bsky.social
· Feb 5

GitHub - grimme-lab/QCxMS2: Program package for the quantum mechanical calculation of EI mass spectra using automated reaction network exploration
Program package for the quantum mechanical calculation of EI mass spectra using automated reaction network exploration - grimme-lab/QCxMS2
github.com
Reposted by Grimme Lab

Reposted by Grimme Lab
Reposted by Grimme Lab

Reposted by Grimme Lab

Reposted by Grimme Lab


Reposted by Grimme Lab
Johannes Gorges
@jogorges.bsky.social
· Jan 17
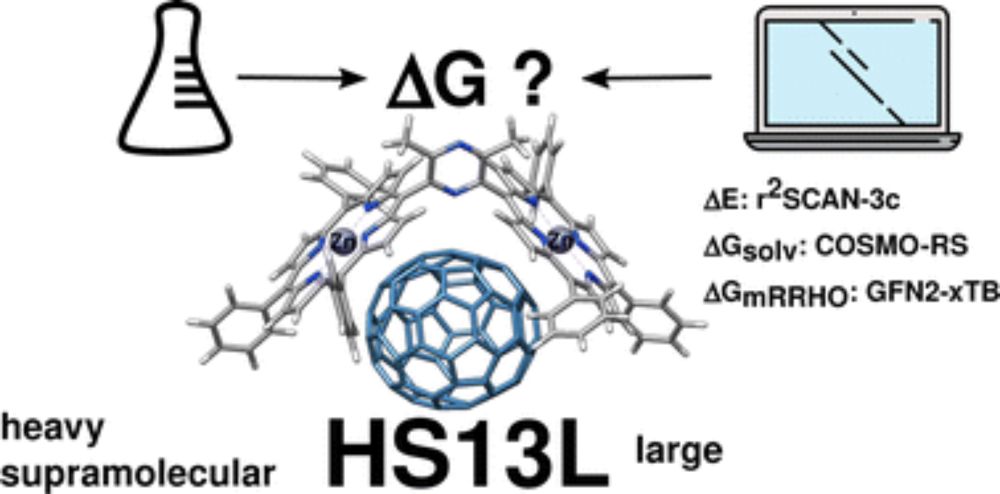
Reliable prediction of association (free) energies of supramolecular complexes with heavy main group elements – the HS13L benchmark set
We introduce a set of 13 supramolecular complexes featuring diverse non-covalent interactions with heavy main group elements (Zn, As, Se, Te, Br, I), high charges (−2 up to +4), and large systems with...
doi.org