
Thomas Froitzheim
@thfroitzheim.bsky.social
110 followers
160 following
3 posts
Theoretical chemist in the @grimmelab.bsky.social working on semiempirical methods and excited states
Posts
Media
Videos
Starter Packs


Reposted by Thomas Froitzheim

Reposted by Thomas Froitzheim

Jan Řezáč
@jrezac.bsky.social
· Jun 26
Reposted by Thomas Froitzheim

Q-Chem
@qchemsoftware.bsky.social
· Jun 26
Reposted by Thomas Froitzheim

Marcel M
@mrclmllr.bsky.social
· Jun 24




Reposted by Thomas Froitzheim
Marcel M
@mrclmllr.bsky.social
· Dec 17
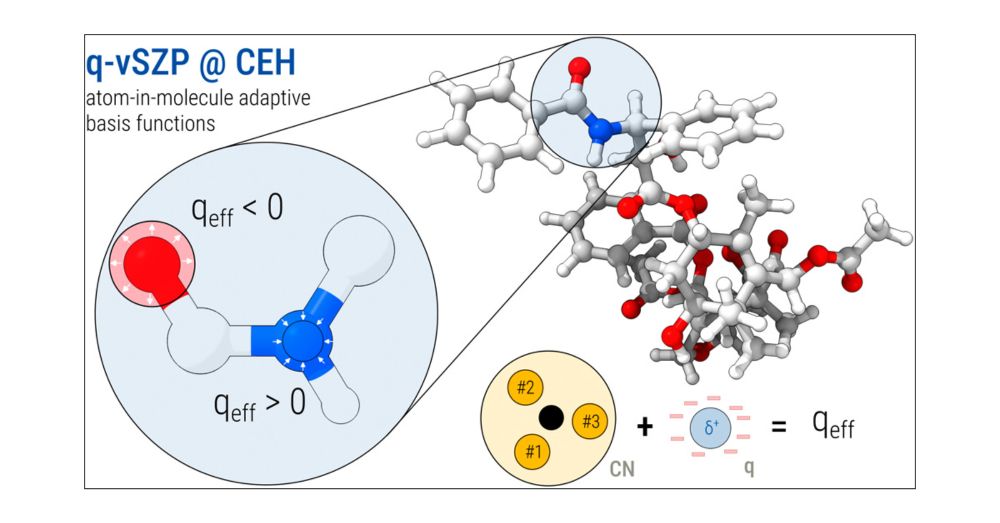
Advanced Charge Extended Hückel (CEH) Model and a Consistent Adaptive Minimal Basis Set for the Elements Z = 1–103
The Charge Extended Hückel (CEH) model, initially introduced for adaptive atomic orbital (AO) basis set construction (J. Chem. Phys. 2023, 159, 164108), has been significantly revised to enhance accur...
pubs.acs.org