Sina Majidian
@sinamajidian.bsky.social
1.3K followers
1.6K following
89 posts
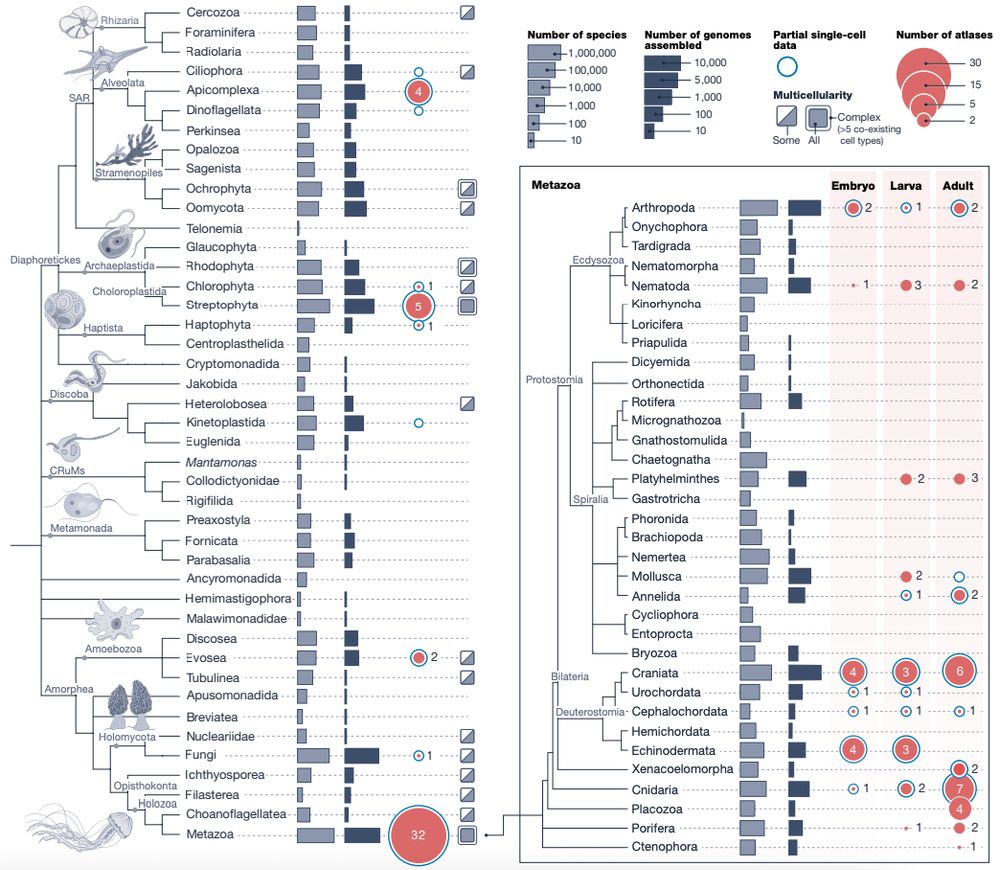
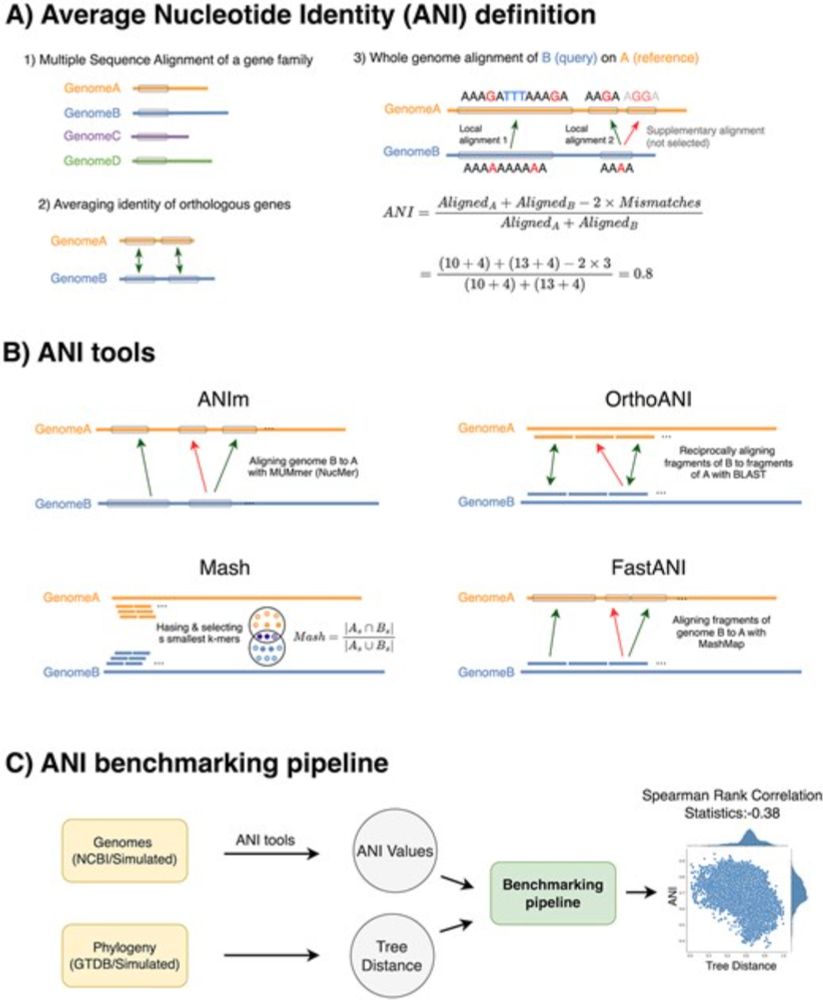
On the academic job market | How are species compared to one another across different genomic regions? Postdoc at Langmead Lab, Johns Hopkins | Comparative #genomics at scale | Formerly at UNIL/SIB/WUR | sinamajidian.github.io
Posts
Media
Videos
Starter Packs
Pinned

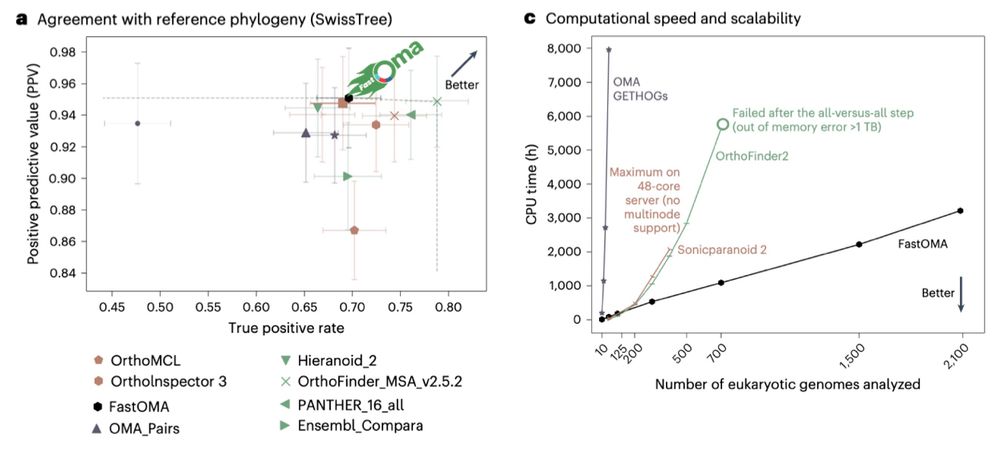
Reposted by Sina Majidian



Reposted by Sina Majidian








Reposted by Sina Majidian


Reposted by Sina Majidian



Reposted by Sina Majidian




Reposted by Sina Majidian



Reposted by Sina Majidian


Reposted by Sina Majidian

Reposted by Sina Majidian





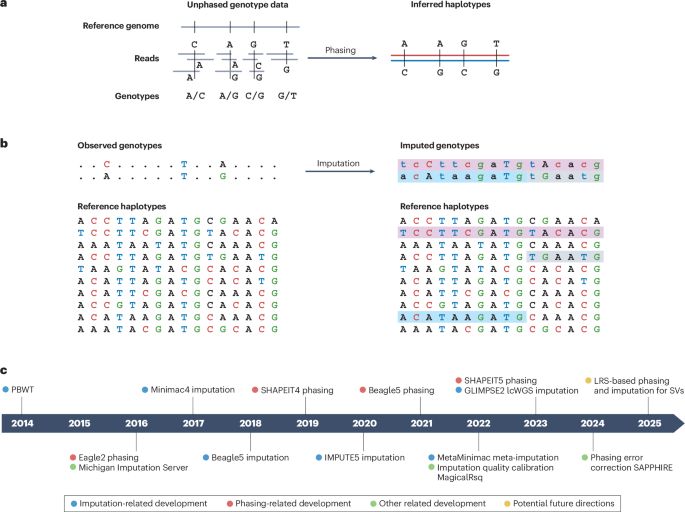
Reposted by Sina Majidian