
Thilo Muth
@drmuth.bsky.social
300 followers
560 following
67 posts
Group Leader and Adjunct Professor 🧑🏫 | Public Health | Data Engineering & Visualization | 🧬 Bioinformatics & 📈 Mass Spec Enthusiast | 📍 Berlin | Passionate About Collaboration and Open Science 🔬✨
Posts
Media
Videos
Starter Packs






Reposted by Thilo Muth




Thilo Muth
@drmuth.bsky.social
· Aug 26
Reposted by Thilo Muth

Daniel Figeys
@dfigeys.bsky.social
· Aug 22

Peptide abundance correlations in metaproteomics enhance taxonomic and functional analysis of the human gut microbiome - npj Biofilms and Microbiomes
npj Biofilms and Microbiomes - Peptide abundance correlations in metaproteomics enhance taxonomic and functional analysis of the human gut microbiome
www.nature.com
Reposted by Thilo Muth

Alison Chaves
@alisonchaves.bsky.social
· Aug 15

PSManalyst: A Dashboard for Visual Quality Control of FragPipe Results
FragPipe is recognized as one of the fastest computational platforms in proteomics, making it a practical solution for the rapid quality control of high-throughput sample analyses. Starting with version 23.0, FragPipe introduced the “Generate Summary Report” feature, offering .pdf reports with essential quality control metrics to address the challenge of intuitively assessing large-scale proteomics data. While traditional spreadsheet formats (e.g., tsv files) are accessible, the complexity of the data often limits user-friendly interpretation. To further enhance accessibility, PSManalyst, a Shiny-based R application, was developed to process FragPipe output files (psm.tsv, protein.tsv, and combined_protein.tsv) and provide interactive, code-free data visualization. Users can filter peptide-spectrum matches (PSMs) by quality scores, visualize protease cleavage fingerprints as heatmaps and SeqLogos, and access a range of quality control metrics and representations such as peptide length distributions, ion densities, mass errors, and wordclouds for overrepresented peptides. The tool facilitates seamless switching between PSM and protein data visualization, offering insights into protein abundance discrepancies, samplewise similarity metrics, protein coverage, and contaminants evaluation. PSManalyst leverages several R libraries (lsa, vegan, ggfortify, ggseqlogo, wordcloud2, tidyverse, ggpointdensity, and plotly) and runs on Windows, MacOS, and Linux, requiring only a local R setup and an IDE. The app is available at (https://github.com/41ison/PSManalyst.
pubs.acs.org
Thilo Muth
@drmuth.bsky.social
· Aug 19

Evaluation of Parallel Accumulation-Serial Fragmentation methods for metaproteomics using a model microbiome
Mass spectrometry-based metaproteomics allows for the identification and quantification of thousands of proteins from clinical and environmental samples and is rapidly gaining importance in microbiome...
doi.org
Reposted by Thilo Muth

Robert Koch-Institut
@rki.de
· Aug 18

Reducing meat consumption: Results from a German survey on attitudes, behaviour and willingness to change among adults
Background Individual meat consumption in Germany has fallen slightly in recent years, but still exceeds the recommended quantities. High meat consumption has negative impacts both on human health and...
journals.plos.org
Reposted by Thilo Muth

Lukas Schwyter
@schluk.bsky.social
· Feb 25

Reposted by Thilo Muth

Albert Heck
@hecklab.bsky.social
· Jun 2

Deep Coverage and Extended Sequence Reads Obtained with a Single Archaeal Protease Expedite de novo Protein Sequencing by Mass Spectrometry
The ability to sequence proteins without reliance on a genomic template defines a critical frontier in modern proteomics. This approach, known as de novo protein sequencing, is essential for applicati...
www.biorxiv.org
Reposted by Thilo Muth


Reposted by Thilo Muth


Reposted by Thilo Muth



Thilo Muth
@drmuth.bsky.social
· Aug 7
Generalizable direct protein sequencing with InstaNexus
Protein-based therapeutics, such as antibodies and nanobodies, are not encoded in reference genomes, challenging their accurate characterization via standard proteomics. Current methods rely on indire...
doi.org
Reposted by Thilo Muth

Mak Saito
@maksaito.bsky.social
· Nov 10
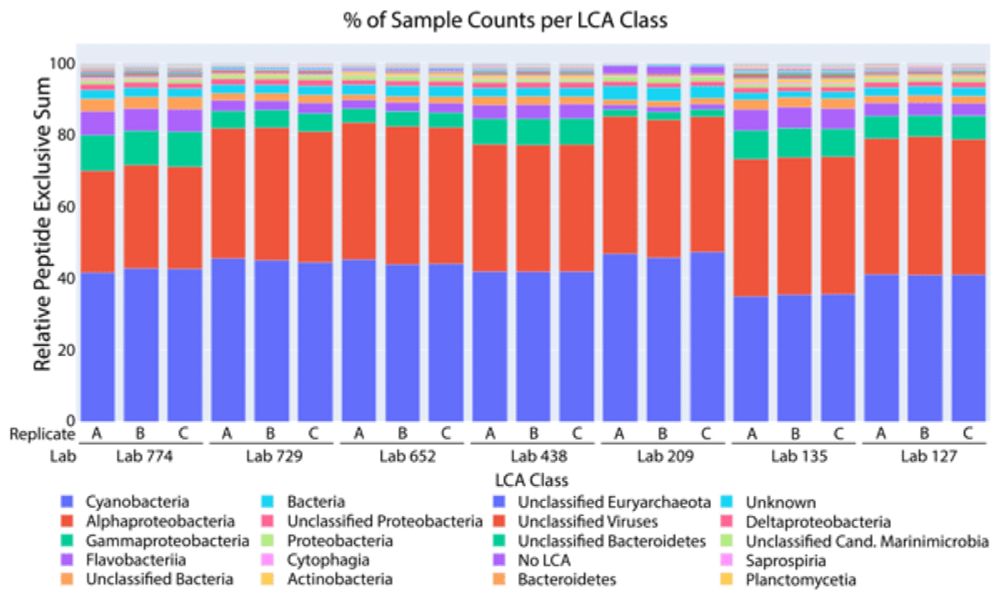
Results from a multi-laboratory ocean metaproteomic intercomparison: effects of LC-MS acquisition and data analysis procedures
Abstract. Metaproteomics is an increasingly popular methodology that provides information regarding the metabolic functions of specific microbial taxa and has potential for contributing to ocean ecolo...
bg.copernicus.org
Thilo Muth
@drmuth.bsky.social
· Aug 2
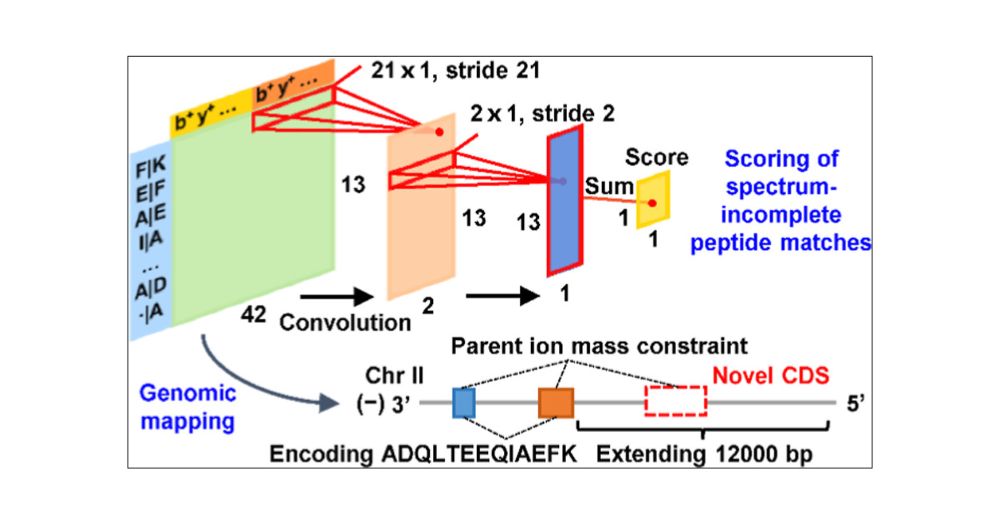
Precise Discovery of Novel N-Terminal Proteoforms beyond the Limitations of Proteogenomics and De Novo Sequencing
Alternative splicing-mediated protein N-terminal sequence variation is closely associated with diseases, but its identification by mass spectrometry faces technical bottlenecks. Traditional proteogeno...
pubs.acs.org
Reposted by Thilo Muth

Reposted by Thilo Muth

Martin Hölzer
@martinhoelzer.bsky.social
· Jul 31
GitHub - hoelzer/dfg: A LaTeX template for a basic DFG (Deutsche Forschungsgemeinschaft, German Research Foundation) grant proposal.
A LaTeX template for a basic DFG (Deutsche Forschungsgemeinschaft, German Research Foundation) grant proposal. - hoelzer/dfg
github.com
Thilo Muth
@drmuth.bsky.social
· Aug 2
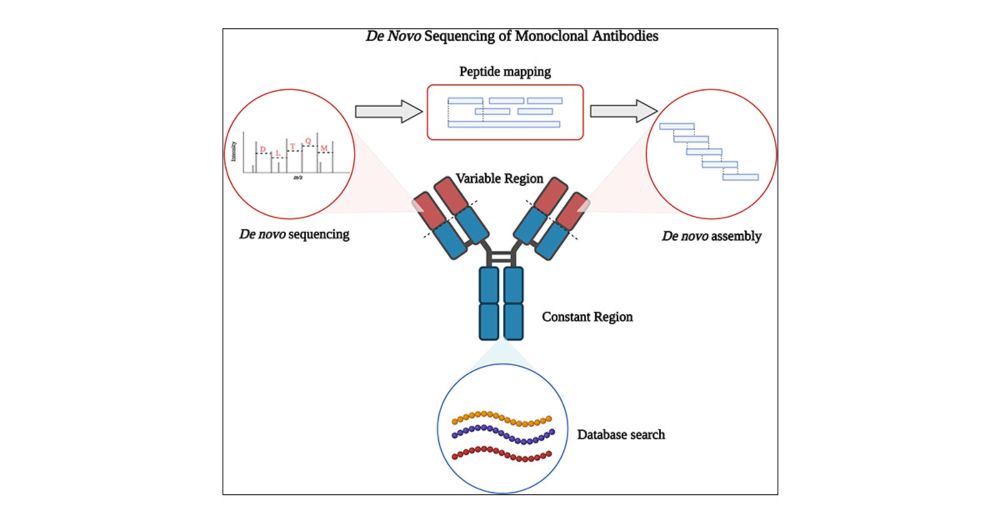
Homo-Tag-Assembler Assay for Full-Length Antibody Sequencing
Antibodies are proteins produced by the immune system in response to antigens. Determining the sequence of purified unknown antibodies remains difficult due to factors like incomplete sequence coverage, ambiguous isobaric residues (I/L), and unstable assembly processes. To overcome these issues, we developed the Homo-Tag-Assembler, a software tool using a homology tag fuzzy string matching algorithm. By analyzing amino acid distributions from many similar antibody variable region sequences, we created a probability table specific to these regions. This helped evaluate peptide similarity during de novo sequencing, distinguish peptides from light and heavy chains, and differentiate leucine from isoleucine. Testing with the Herceptin antibody showed full-length sequence coverage with accuracy of 99.1% for the light chain and 100% for the heavy chain variable regions. This method uses conserved regional features and homology based information to enable accurate full-length antibody sequence reconstruction.
pubs.acs.org