Xi Fu
@fuxialexander.bsky.social
110 followers
450 following
28 posts
Transcription regulation; deep learning; (bad) developer
Posts
Media
Videos
Starter Packs



Xi Fu
@fuxialexander.bsky.social
· May 5
Reposted by Xi Fu

Reposted by Xi Fu

Sara Mostafavi
@saramostafavi.bsky.social
· Apr 16

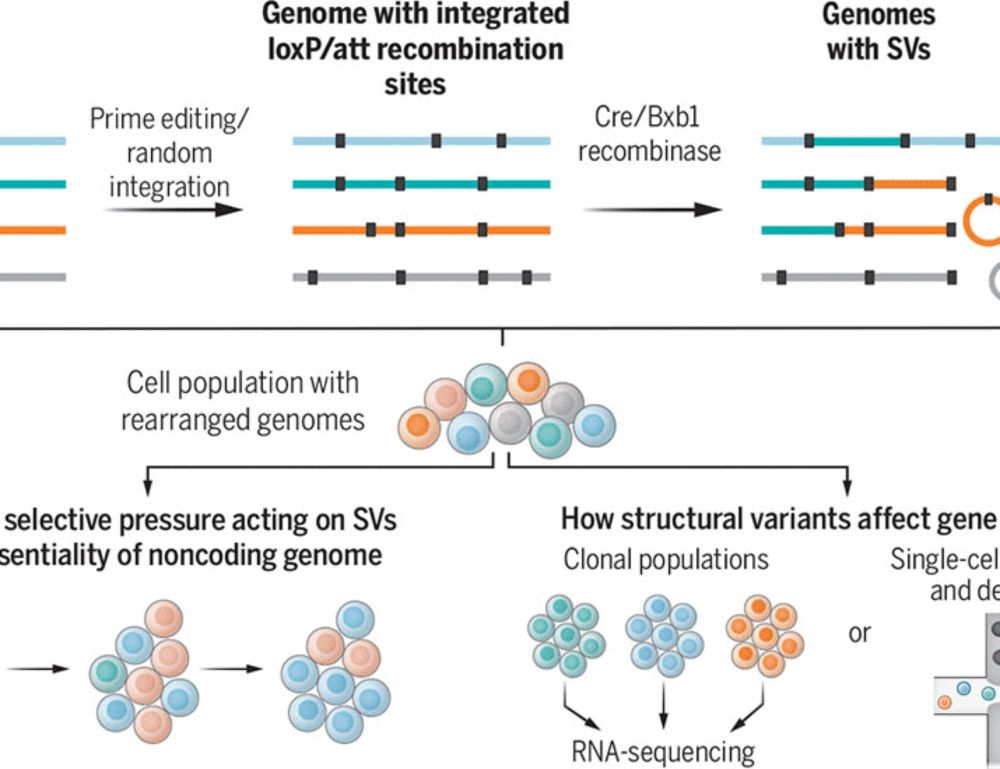
Deep genomic models of allele-specific measurements
Allele-specific quantification of sequencing data, such as gene expression, allows for a causal investigation of how DNA sequence variations influence cis gene regulation. Current methods for analyzin...
www.biorxiv.org
Reposted by Xi Fu



Reposted by Xi Fu

Xi Fu
@fuxialexander.bsky.social
· Mar 21
Xi Fu
@fuxialexander.bsky.social
· Mar 18
Reposted by Xi Fu

Reposted by Xi Fu

Reposted by Xi Fu

Reposted by Xi Fu
Tuuli Lappalainen
@tuuliel.bsky.social
· Feb 12

Gamze Gürsoy
@gamzeandgursoy.bsky.social
· Feb 12

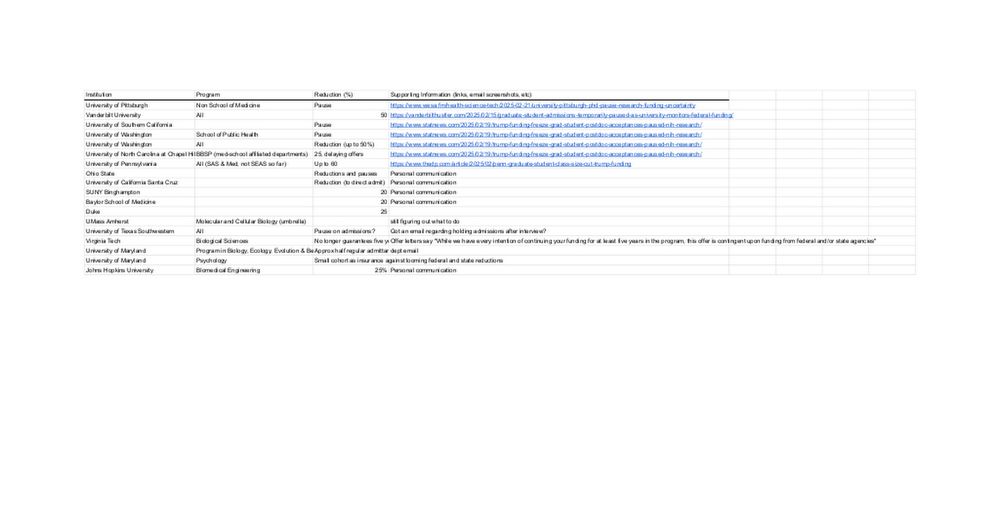
Secure and federated quantitative trait loci mapping with privateQTL
Choi et al. developed a novel tool for privacy-preserving cross-institutional eQTL
mapping studies. The authors benchmarked their tool against meta-analysis and demonstrated
that it achieves higher ac...
www.cell.com
Xi Fu
@fuxialexander.bsky.social
· Feb 12

Xi Fu
@fuxialexander.bsky.social
· Feb 12
Xi Fu
@fuxialexander.bsky.social
· Feb 12
Xi Fu
@fuxialexander.bsky.social
· Feb 12


Reposted by Xi Fu

Reposted by Xi Fu

Peter Koo
@pkoo562.bsky.social
· Feb 5
Xi Fu
@fuxialexander.bsky.social
· Feb 5
Xi Fu
@fuxialexander.bsky.social
· Feb 5
Xi Fu
@fuxialexander.bsky.social
· Feb 1